Cholesterol-dependent cytolysins (CDCs) are key virulence factors involved in many lethal bacterial infections, including pneumonia, necrotizing soft tissue infections, bacterial meningitis, and miscarriage. Host responses to these diseases involve myeloid cells, especially macrophages. Macrophages use several systems to detect and respond to cholesterol-dependent cytolysins, including membrane repair, mitogen-activated protein (MAP) kinase signaling, phagocytosis, cytokine production, and activation of the adaptive immune system. However, CDCs also promote immune evasion by silencing and/or destroying myeloid cells. While there are many common themes between the various CDCs, each CDC also possesses specific features to optimally benefit the pathogen producing it.
- pore-forming toxin
- streptolysin
- perfringolysin
Note:All the information below can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Cholesterol-dependent cytolysins (CDCs) are a subset of pore-forming toxins that serve as key virulence factors for a wide range of lethal and opportunistic Gram-positive bacterial pathogens that collectively infect or invade nearly all parts of the human body. Consequently, hosts attempt to eliminate these pathogens with both general and tissue-specific approaches. One common approach that has tissue-specific flexibility is activation and polarization of macrophages. Macrophages coordinate the local tissue response with cytokines and can directly eliminate bacteria through phagocytosis and secretion of reactive oxygen/nitrogen species. They further promote wound repair and restore the tissue to homeostasis. As a result, pathogenic bacteria target macrophages for elimination, reprogramming, or shelter. CDCs figure prominently in many of these attempts. This review explores both general and specific molecular mechanisms used by CDCs to kill, control, or evade macrophages.
2. CDC Interactions with Macrophages
2.1. Cytokine Production in Response to CDCs
One key consequence of activating the signaling pathways, especially the p38 pathway, is production of pro-inflammatory cytokines. The major pro-inflammatory cytokines induced by CDC challenge are Tumor Necrosis Factor α (TNFα,) Interleukin (IL)-1β, IL-6, and IL-8, though other cytokines and chemokines are also CDC-dependent. However, one challenge with CDC-dependent cytokine production is ascertaining if the cytokine is produced by signaling pathways directly stimulated by the CDC or indirectly by other danger- or pathogen-associated molecular patterns (DAMPs and PAMPs). Many studies attributing cytokine production to CDCs used bacteria with and without deletion of the CDC. Since CDCs are often key to bacterial virulence, it is difficult to discern if changes in pro-inflammatory cytokine production are due to overall reduced virulence or directly due to the toxin. Pure CDCs may contain contaminants, such as toll-like receptor (TLR) ligands, which complicate interpretation of data. Often studies do not include inactive toxins to control for the presence of any contaminants in purified toxins. Cellular damage by CDCs further releases several DAMPs, including IL-1α, ATP, and high mobility group box 1 protein (HMGB1) [1][2][3][4][111–114], which may exert autocrine and paracrine effects on cells, including TLR engagement and pro-inflammatory cytokine production. These effects may be especially apparent when longer (12+ h) time points are used. Finally, signaling pathways can be activated independently of plasma membrane receptors by the Ca2+ fluctuations that occur during membrane damage and repair [5][47]. Thus, extreme care should be taken in interpreting pro-inflammatory cytokine production in response to CDCs.
Direct cytokine production in response to CDCs is triggered by inflammasome (see Section 2.2), p38, and Ca2+ dependent pathways. Activation of p38 by SLO, PLY, ALO, and VLY leads to IL-8 production [6][7][8][22,73,77]. Activation of p38 also induces secretion of macrophage migration inhibitory factor (MIF) by PLY, which helps reduce the bacterial load [9][83]. Finally, p38 also stimulates TNFα production after SLO challenge [10][115]. TNFα is critical for recruiting macrophages during a subcutaneous S. pyogenes infection, which limits bacterial dissemination [11][116]. CDCs also stimulate IL-6 production. LLO, PLY, and SLY stimulate IL-6, which is Ca2+ dependent for LLO and PLY [12][13][14][15][117–120]. Similarly, LLO and PLY trigger IL-1α release and calpain activation in a Ca2+ dependent fashion [3][4][113,114]. Overall, CDCs can activate pro-inflammatory cytokine signaling.
The ability of CDCs to directly stimulate TLR signaling pathways remains controversial. While some studies suggest that PLY [16][17][121,122], ALO, LLO, SLO, and PFO [18][123] all trigger TLR4, other studies have not observed TLR4-dependent responses [19][20][124,125]. Similarly, NF-ΚB activation, which is downstream of TLRs, is variably reported for CDCs. Some studies observe NF-ΚB activation by CDCs [21][22][126,127], while another did not [20][125]. One potential explanation for the discrepancy is the autocrine and the paracrine effects involving TLRs and/or IL-1 receptor (IL-1R), which also signals through MyD88. LLO-induced NF-ΚB is IL-1R dependent [23][128]. PLY-induced TNFα production and TLR4 activation were measured at 24 h [24][25][129,130], thus TLR4 activation could occur secondary to DAMP release. There are also cell type differences in cytokine production in response to CDCs. For example, PLY induces opposite effects for TNFα production in dendritic cells and macrophages [26][131]. Overall, TLR activation may be secondary to other effects of CDCs.
2.2. CDCs Activate the Inflammasome
Along with the previously discussed cytokines, CDCs promote IL-1 secretion [25][27][28][130,138,139]. IL-1β is usually secreted following activation of the inflammasome. The inflammasome is a multiprotein complex comprising a cytoplasmic sensory pattern-recognition receptor, the scaffolding protein apoptosis-associated speck-like protein containing a CARD (ASC)/Pycard and an inflammatory caspase (caspase-1 or caspase-11 in mouse, caspases-1,4 or 5 in human) that promotes the inflammatory cell death process pyroptosis and the activation/release of pro-inflammatory cytokines IL-1β and IL-18 (reviewed in [1][29][111,140]) (Figure 1). Sensory pattern-recognition receptors that activate the inflammasome include proteins in both the Pyhin-family and the nod-like receptors (NLRs). They sense various pathogen-associated molecular patterns and danger-associated molecular patterns, including membrane perforation by CDCs [1][29][111,140]. Membrane perforation is sensed by nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3), presumably indirectly via loss of K+ [30][141], while cytoplasmic bacterial or mitochondrial DNA is sensed by absent in melanoma 2 (AIM2) [31][142] (Figure 1). After activation, NLRP3 or AIM2 oligomerize ASC, which recruits and activates caspase-1. Activated caspase-1 then cleaves the pore-forming toxin gasdermin D to promote pyroptosis (Figure 1). Pyroptosis prevents intracellular pathogens from sheltering in the cell and releases pro-inflammatory mediators, including HMGB1, IL-1β, and IL-18 [1][29][111,140]. The inflammasome promotes anti-pathogen responses after sensing the CDC challenge.
CDCs activate two inflammasomes. CDCs directly activate the NLRP3 inflammasome via membrane perforation and K+ efflux, and some also indirectly activate AIM2 by facilitating the entry of mitochondrial or bacterial DNA into the cytosol. PFO [32][143], SLO [33][34][37,144], TLO [35][125], PLY [36][37][102,145], LLO [38][39][40][146–148], and SLY [41][149] all activate the NLRP3 inflammasome. LLO-mediated phagosomal rupture and lysosomal permeabilization further activate NLRP3 [39][147]. Bacteria deficient in LLO [40][42][148,150], PLY [36][37][102,145], PFO [32][143], and SLO [34][144] fail to stimulate IL-1β production. Other toxins, including streptolysin S [43][69] and C. perfringens α-toxin [32][143], fail to activate the inflammasome. Thus, CDCs are necessary for pathogen sensing by the NLRP3 inflammasome.
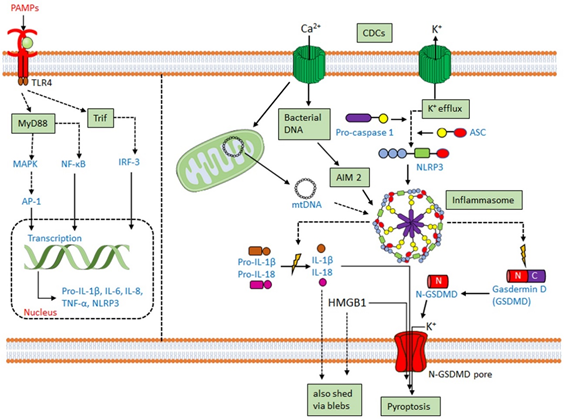
AIM2 activation has functional redundancy with NLRP3 in responding to CDCs. AIM2 can be activated by either bacterial DNA or mitochondrial DNA following L. monocytogenes infection or PLY intoxication [44][45][46][47][48][70,84,151–153]. AIM2 is also activated by mitochondrial DNA released into the cytosol after cholesterol perturbations [49][154]. Upregulation of the enzyme cholesterol-25-hydroxylase (Ch25h) by type I or type II interferons (IFNs) produces the regulatory oxysterol, 25-hydroxy-cholesterol [50][155]. The 25-hydroxy-cholesterol regulates cholesterol biosynthesis, flux, and storage [51][52][156,157]. Deletion of Ch25h from macrophages leads to increased IL-1β production in response to LPS-mediated type I interferon (IFN) induction [53][158]. When IFNs are unable to induce Ch25h, macrophages undergo cholesterol overload and switch to aerobic glycolysis with mitochondrial damage [49][154]. Mitochondrial damage permits the escape of mitochondrial DNA into the cytosol, activating the AIM2 inflammasome to overproduce IL-1β [49][154]. Consequently, Ch25h deletion confers AIM2-dependent protection from L. monocytogenes infection on macrophages [49][54][154,159].
2.3. CDCs Damage Phagosomes and Permit Phagolysosomal Escape
Phagocytosis is the cellular engulfment process of large particles (>0.5 µm in diameter). It is used by innate immune cells such as macrophages, dendritic cells, and neutrophils to internalize and kill extracellular pathogens [55][167]. Intracellular pathogens typically hijack phagocytosis to prevent phagosome fusion with the lysosome and may escape into the cytosol. Indeed, L. monocytogenes requires LLO for phagosomal escape and for preventing fusion with lysosomes [56][57][58][168–170]. While it is known that transgenic expression of other CDCs such as PFO can promote the escape of L. monocytogenes [59][171] or Bacillus subtilis [60][61][172,173], it is now appreciated that other traditionally extracellular bacteria such as S. pyogenes and C. perfringens rely on CDC-dependent phagosome interference to promote infection [62][63][64][65][174–177]. However, phagosomal escape is not perfect [66][178], thus it is not an all-or-none process. Interestingly, CDCs variably stimulate or impair phagocytosis. LLO stimulates ion flux, which promotes the internalization of L. monocytogenes [67][68][179,180]. Conversely, SLO interferes with phagocytosis [69][181]. However, after membrane damage and repair, compensatory endocytosis is activated to restore homeostasis [70][71][36,182]. Before phagosomal escape, LLO, PFO, and PLY may also interfere with acidification in non-macrophages [57][169]. However, lysosomal membrane permeability stimulates inflammasome activation in macrophages [72][73][74][183–185]. Consequently, when PLY interferes with lysosomal acidification in macrophages, it also drives cell death [75][186]. Interestingly, these events appear to occur independently of phagosome escape. Importantly, in macrophages, LLO permeabilizes phagosomes to small molecules prior to large ones, which led the authors to conclude that LLO forms pores of different sizes, possibly due to insertion of incomplete pores [58][170]. However, ESCRT-III mediates phagosomal membrane repair [76][187], thus an alternative explanation is that repair mechanisms limit the extent of damage caused by LLO, preventing the loss of larger molecules. While the structure of LLO is optimized for activity at low pH [77][188], the host protein gamma-interferon inducible lysosomal thiolreductase (GILT) further activates LLO by reducing the single cysteine in the protein [78][189]. Thus, CDCs extensively target phagocytosis to evade cell death and promote escape and immune evasion.
2.4. CDC-Mediated Innate Immune Evasion
While CDCs activate several cell defense mechanisms, they also contribute to evading immune activation. Immune evasion is best described for SLO and PLY. CDCs interfere with phagocytosis and inhibit cytokine production. SLO reduces phagocytosis and S. pyogenes killing by neutrophils [69][181]. Similarly, PLY-stimulated pyroptosis of neutrophils lead to elastase release, which blocks phagocytosis of S. pneumoniae in the inflammasome-defective macrophage cell line Raw264.7 [79][42]. SLO may stimulate the ubiquitination and the degradation of IL-1β [80][190]. Similarly, PLY expression reduces maturation of human DCs and pro-IL-1β, IL-8, and IL-12p70 production in response to S. pneumoniae [81][191]. Finally, PLY, PFO, and SLO can block TNFα production [19][26][124,131]. Thus, CDCs promote immune evasion.
2.5. CDCs as an Adaptive Immune Target
While CDCs try to impair macrophages, CDCs are popular targets for antigen presentation by macrophages and other APCs to the adaptive immune system. Bacteriocidal T and B cell responses to LLO are mounted against L. monocytogenes [82][83][200,201], LLO-expressing Escherichia coli [84][202], or B. subtilis [85][203]. Memory CD4+ T cells respond to PLY [86][204] and are associated with an absence of S. pneumoniae carriage [87][205]. Antibodies to ALO [88][206], PFO [89][207], or SLY [90][12] protect mice from lethal infections. Antibodies to CDCs are readily produced [13][82][88][91][92][93][118,200,206,208–210]. Anti-SLO and anti-PLY titers can be detected in human serum [93][94][210,211], indicating CDCs are robustly antigenic in humans. Importantly, anti-CDC T and B cell responses do not depend on hemolytic activity [92][95][209,212], which suggests that non-hemolytic CDC toxoids may serve as useful vaccine targets.
This excerpt is from Thapa et al Toxins (Basel). 2020 Aug 19;12(9):E531. Refer to the article for reference numbers and more information.