 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Koji Hatano | + 2176 word(s) | 2176 | 2020-08-21 05:56:59 | | | |
| 2 | Camila Xu | Meta information modification | 2176 | 2020-08-25 04:51:49 | | | | |
| 3 | Camila Xu | Meta information modification | 2176 | 2020-08-27 11:23:01 | | |
Video Upload Options
Chronic inflammation is a major cause of human cancers. The environmental factors, such as microbiome, dietary components, and obesity, provoke chronic inflammation in the prostate, which promotes cancer development and progression. Crosstalk between immune cells and cancer cells enhances the secretion of intercellular signaling molecules, such as cytokines and chemokines, thereby orchestrating the generation of inflammatory microenvironment. Tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) play pivotal roles in inflammation-associated cancer by inhibiting effective anti-tumor immunity. Anti-inflammatory agents, such as aspirin, metformin, and statins, have potential application in chemoprevention of prostate cancer. Furthermore, pro-inflammatory immunity-targeted therapies may provide novel strategies to treat patients with cancer. Thus, anti-inflammatory agents are expected to suppress the “vicious cycle” created by immune and cancer cells and inhibit cancer progression. This review has explored the immune cells that facilitate prostate cancer development and progression, with particular focus on the application of anti-inflammatory agents for both chemoprevention and therapeutic approach in prostate cancer.
1. Introduction
Chronic inflammation plays a major role in the etiology and development of various types of malignant tumors, including hepatocellular carcinoma, gastric cancer, lung cancer, colorectal cancer, and prostate cancer [1][2][3][4][5]. Although inherited germline mutations are involved in prostate cancer development [6][7], immigration studies indicate the importance of environmental factors; for instance, it was found that immigrants from Asian countries in Western countries acquired higher prostate cancer risks within one generation [8][9]. The exposure to environmental factors, such as microbiome, cellular trauma, hormonal imbalances, dietary carcinogens, and obesity, leads to prostate epithelium injury and causes chronic inflammation [3][10][11][12]. In the adult prostate, chronic inflammation is prevalent and associated with putative precursor lesions that can provoke prostate cancer development [13][14][15][16].
A meta-analysis revealed an increased risk of prostate cancer among men with a history of prostatitis, syphilis, and gonorrhea [17]. Although a number of studies supported the idea of a connection between prostatitis and prostate cancer risk [18][19][20][21][22], subsequence studies revealed conflicting results [23][24][25][26][27]. The inconsistent results could be attributed to differences in the study population and potential selection bias, as acute and chronic prostatitis is associated with increased serum prostate-specific antigen (PSA) levels [28]. Previous epidemiological studies focused on the relationship between inflammation and prostate cancer development [29][30][31]. The first prospective study in men without biopsy indication revealed that benign tissue inflammation was positively associated with prostate cancer development [32]. Furthermore, the progression and aggressiveness of prostate cancer was reportedly associated with systemic inflammation markers in the serum, such as C-reactive protein levels, as well as differential blood cell count (neutrophils, lymphocytes, monocytes, and platelets) [33][34][35][36][37].
The pathogenesis of inflammation-associated cancer is complex as both the innate and adaptive immune systems are involved in the process [38][39][40]. Chronic inflammation in the prostate microenvironment causes chronic increase in reactive oxygen species, which is associated with oxidative DNA damage in prostate epithelium [41]. Accumulated DNA damage can cause somatic mutations in key tumor suppressor genes and induce genome instability resulting in genomic changes in oncogenes, thus facilitating the development and progress of prostate cancer [3][42][43]. In fact, in patients with castration-resistant prostate cancer (CRPC), aberrations of AR (androgen receptor), E26 transformation-specific genes, TP53 (tumor protein p53), and PTEN (phosphatase and tensin homolog) were frequently observed (40–60% of cases), whereas only few (8%) cases had pathogenic germline alterations [44]. Additionally, inflammatory cells also secrete cytokines and chemokines that stimulate prostate cancer growth, angiogenesis, invasion, and metastasis [45][46][47]. Thus, anti-inflammatory agents are expected to suppress inflammation in the tumor microenvironment and inhibit prostate cancer progression (Figure 1).
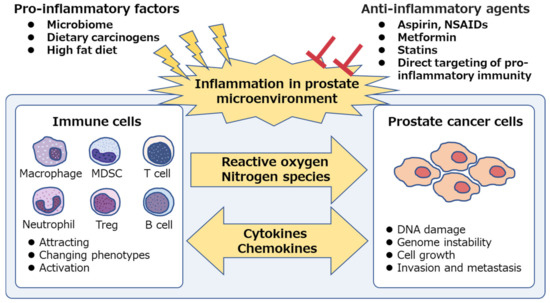
Figure 1. Chronic inflammation is associated with prostate cancer. Pro-inflammatory factors, such as microbiome and dietary components, are the potential cause of prostatic inflammation. Immune cells secrete reactive oxygen and nitrogen species, induce DNA damage and genome instability in prostate epithelium and cause prostate cancer development. Both immune cells and prostate cancer cells secrete intercellular signaling molecules, such as cytokines and chemokines, and contribute to the generation of inflammatory microenvironment, which facilitates cancer progression. Anti-inflammatory agents suppress the “vicious cycle” and inhibit prostate cancer development and progression. NSAIDs, non-steroidal anti-inflammatory drugs; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell.
2. Immune Cells Involved in Inflammation and Prostate Cancer Progression
Cancer development and its response to therapy are strongly affected by innate and adaptive immunity, which either promote or attenuate tumorigenesis and can have opposing effects on therapeutic outcome [48]. A large number of studies have focused on immune cells in prostate cancer, including innate immune cells: Macrophages, neutrophils, and mast cells; adaptive immune cells: T cells and B cells; and immune-suppressive cells: regulatory T cells (Treg cells) and myeloid-derived suppressor cells (MDSCs) [49]. In the prostate microenvironment, these immune cells act as either friends or foes [50]. Growing evidence suggests that both macrophages and MDSCs play pivotal roles in inflammation-associated prostate cancer development and progression via down-regulation of effective anti-tumor immunity (Figure 2). Although the precise molecular mechanisms involved are still unclear, animal models of prostate cancer, which mimic the human disease, have contributed to the determination of specific pathways and helped develop novel therapeutic agents.
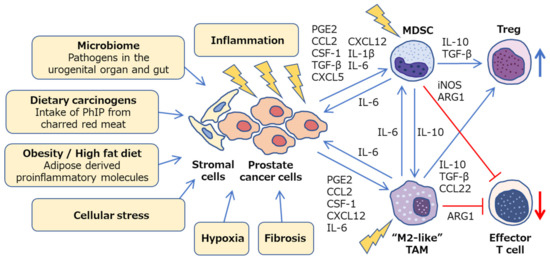
Figure 2. Inflammatory microenvironment in prostate cancer. Diverse mechanisms, such as microbiome, dietary carcinogens, obesity, cellular stress, hypoxia and fibrosis, can be a potential cause of inflammation in prostate cancer. Crosstalk between cancer cells, stromal cells, and immune cells promotes chronic inflammation and facilitates prostate cancer progression via a variety of intercellular signaling molecules. Prostate tumor microenvironment is generally considered to be immunologically “cold” as M2-like TAMs and MDSCs cooperatively inhibit effector T cells and activate regulatory T cells. PhIP, 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine; PGE2, prostaglandin E2; CSF-1, colony stimulating factor-1; TGF-β, transforming growth factor-β; ARG1, arginase 1; iNOS, inducible nitric oxide synthase; TAM, tumor-associated macrophage; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell.
2.1. Macrophages
Tumor-associated macrophages (TAMs) are crucial drivers of tumor promoting inflammation and are generally associated with poor prognosis in solid tumors [51], including prostate cancer [52][53][54][55][56][57][58]. TAMs contribute to tumor progression at different levels by promoting genetic instability, cell proliferation, angiogenesis, and metastasis, as well as suppressing protective adaptive immunity [59][60][61][62][63]. Historically, macrophages are divided into classically activated macrophages (M1) and alternatively activated macrophages (M2), exhibiting anti-tumoral and pro-tumoral properties, respectively. Although interleukin (IL)-4 and IL-13 are the acknowledged signals that regulate M2 polarization of macrophages, recently other subtypes of the M2 class have been identified [64][65][66][67]. Thus, TAM population is referred to as “M2-like” when they include diverse phenotypes that share the functional outputs of tumor promotion and adaptive immunity suppression; these typically express characteristic surface molecules, such as CD163, CD204, and CD206 [62].
Furthermore, elements within the tumor microenvironment, such as hypoxia, fibrosis, cellular stress, and inflammation, dramatically shift the macrophage polarity towards M2-like phenotypes [68][69][70] (Figure 2). Many studies have shed light on the complex signaling network that drives myeloid cells toward M2-like TAMs, which involves various cytokines, chemokines, and signals within the tumor microenvironment [71], including prostaglandin E2 (PGE2) [72], chemokine (C–C motif) ligand (CCL)2 [73], colony stimulating factor (CSF)-1 [74], C–X–C motif chemokine (CXCL)12, and IL-6 [75][76]. Thus, tumor microenvironment can influence TAM polarization by releasing various factors that give rise to a large spectrum of pro-tumoral TAMs [77]. TAMs inhibit effector T cells by secreting IL-10, transforming growth factor (TGF)-β, and arginase (ARG)1 as well as via direct cell to cell contact. They also induce Treg cells via IL-10 and TGF-β [78][79] (Figure 2). Therefore, TAMs can be potential therapeutic targets, and re-education of pro-tumoral M2-like TAMs toward anti-tumorigenic phenotype may be a potent strategy to treat prostate cancer [80][81].
2.2. MDSCs
MDSCs are a tolerogenic and immune-suppressive population of myeloid cells that are significantly expanded in patients with various types of cancers. High MDSC number negatively correlates with disease progression and overall survival, thus suggesting these to be a possible target for cancer immunotherapy [82][83]. MDSCs have distinct phenotypic surface markers as well as functional characteristics, particularly T cell activity inhibition. MDSCs were originally identified in mice as Gr-1+ CD11b+ cells [84][85]. The Gr-1 marker is not a singular molecule, but a combination of Ly6C and Ly6G markers. Currently, MDSCs include two major subsets based on their phenotypic and morphological features: Polymorphonuclear (PMN)-MDSCs, including CD11b+ Ly6Clo Ly6G+ cells, and monocytic (M)-MDSCs, including CD11b+ Ly6Chi Ly6G− cells. In humans, PMN-MDSC equivalent cells are defined as CD11b+ CD14− CD15+ or CD11b+ CD14− CD66b+ and M-MDSCs as CD11b+ CD14+ HLA-DRlow/− CD15− [86]. Growing evidence suggests that MDSCs play an important role in cancer development and progression via suppression of anti-tumoral T cell function in patients with prostate cancer [87][88][89][90][91][92]. However, the MDSC subsets that have clinical relevance during disease progression remain to be identified.
To date, various signaling pathways, including PGE2 [93], CCL2 [94][95], CSF-1 [96], TGF-β [97], CXCL5, CXCL12 [98], IL-1β [99][100], and IL-6 [101], have been identified to be involved in the infiltration and activation of MDSCs in tumor microenvironment [102][103][104][105][106]. MDSCs suppress anti-tumor immunity through a variety of diverse mechanisms, including ARG1, inducible nitric oxide synthase (iNOS), TGF-β, and IL-10, and PMN-MDSCs and M-MDSCs exhibit different mechanisms of immune suppression [107][108][109][110][111][112]. Ultimately, MDSCs inhibit the activation and clonal expansion of tumor-specific T cells, as well as induce Treg cell development (Figure 2). Furthermore, MDSCs secrete various factors that promote prostate cancer progression, such as IL-1 receptor antagonist (IL-1RA) that antagonizes senescence of prostate cancer in a paracrine manner [113], and IL-23 that acts as a driver of CRPC [114]. Thus, targeting MDSCs may provide novel opportunities for cancer therapy.
2.3. Crosstalk between Immune Cells, Stromal Cells, and Cancer Cells in Prostate Microenvironment
In the inflammatory prostate microenvironment, crosstalk between immune, stromal, and cancer cells potentially facilitates further tumor progression [115][116][117]. An ex vivo prostate tumor model, derived from patients with prostate cancer, demonstrated that prostate tumors showed low levels of cytotoxic T lymphocytes and T-helper (Th)1 cells-recruiting chemokines, such as CCL5, CXCL9, and CXCL10, but expressed high levels of chemokines implicated in attracting TAMs, MDSCs, and Treg cells, such as CCL2, CCL22, and CXCL12 [118]. CCL22, secreted by tumor cells and TAMs, activates trafficking of Treg cells within the tumor as well as promotes tumor migration and invasion [119][120][121]. Human prostate carcinoma-associated fibroblasts and prostate cancer cells orchestrate and enhance TAM and MDSC recruitment to prostate tumors as well as M2-like TAM differentiation by the chemokines CCL2, CXCL12, and IL-6 during cancer progression [76][122]. Sexually transmitted disease-associated inflammation facilitates IL-6 production by prostate epithelial cells, which induces M2-like TAM polarization [123]. Furthermore, MDSC and tumor cell cross-talk enhances IL-6 production within tumor microenvironment [124], while IL-10, produced by MDSCs, increases M2-like TAMs [125][126][127]. The MDSCs derived from patients with prostate cancer inhibit CD8+ T cells through ARG1, a downstream signal transducer and activator of transcription (STAT)3 target gene [91]. Moreover, the phenotypic analysis of prostate infiltrating lymphocytes, derived from patients with prostate cancer, revealed them to be skewed towards a regulatory Treg and Th17 phenotypes [128]. Tregs are associated with poor prognosis and were found to be highly infiltrated in the prostate tissue of patients with prostate cancer [129][130]. Th17 cells, the key mediators in a number of autoimmune diseases, play a role in inflammation-associated prostate cancer [131][132]. Their development depends on the pleiotropic cytokine TGF-β, which is also linked to Treg cell development and function [133]. In Hi-Myc mouse model of prostate cancer, retrograde urethral instillation of CP1, a human prostatic isolate of Escherichia coli, was reported to induce chronic inflammation characterized by an influx of TAMs and Th17 lymphocytes with distinct cytokine profiles and thus accelerate cancer progression [134].
Crosstalk between MDSCs and mast cells was found to further suppress effective anti-tumor immunity in a transgenic adenocarcinoma of the mouse prostate (TRAMP) model [135]. PhIP (2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine), one of the most abundant heterocyclic amines in cooked meat, induced rat prostate cancer with elevated DNA mutation frequencies in the prostate as well as infiltration of TAMs and mast cells, suggesting a potential mechanism involving inflammation promotion by which dietary compounds can increase cancer risk [136]. Furthermore, bacterial prostatitis accelerates PhIP-induced prostate carcinogenesis by increasing the level of circulating IL-6 in the rat prostate [137]. Another factor that facilitates chronic inflammation is obesity, wherein adipose derived proinflammatory molecules activate TAMs and MDSCs, subsequently promoting cancer progression [138][139][140][141][142][143][144][145]. CCL2 produced by adipocytes enhances the growth and invasion of prostate cancer cells [146]. Upregulation of serum CCL2 levels enhanced the tumor growth of prostate cancer LNCaP xenografts in high-fat diet fed mice [147]. In obese mice, expanded MDSCs suppress CD8+ T cells via iNOS and interferon-γ, and also induce M2 TAM polarization via IL-10 [148][149]. The loss of Pten in the prostate epithelium causes local MDSC expansion via inflammatory cytokines, such as CSF-1 and IL-1β. [96]. In a mouse prostate cancer model driven by loss of Pten and Smad4, MDSCs play a critical role in cancer progression, as CXCR2-expressing MDSCs infiltrate in the prostate due to CXCL5 up-regulation in tumors [150]. In Pten-deficient model mice for prostate cancer, a high-fat diet mediated inflammation-induced M2 TAM differentiation and expansion of MDSCs, accelerated IL-6 secretion, and facilitated tumor growth via IL-6/STAT3 signaling pathway [151]. Thus, the inflammation-associated prostate cancer progression is potentially mediated by diverse mechanisms, such as microbiome, dietary carcinogens, obesity, cellular stress, hypoxia, and fibrosis, which consequently inhibit effective anti-tumor immunity (Figure 2). These pathways may be promising targets for chemoprevention and cancer therapy.
References
- Ames, B.N.; Gold, L.S.; Willett, W.C. The causes and prevention of cancer. Proc. Natl. Acad. Sci. USA 1995, 92, 5258–5265. [Google Scholar] [CrossRef] [PubMed]
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Intern. Med. 2000, 248, 171–183. [Google Scholar] [CrossRef] [PubMed]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Santi, R.; Tamanini, I.; Galli, I.C.; Perletti, G.; Bjerklund Johansen, T.E.; Nesi, G. Current Knowledge of the Potential Links between Inflammation and Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 3833. [Google Scholar] [CrossRef]
- Leongamornlert, D.; Saunders, E.; Dadaev, T.; Tymrakiewicz, M.; Goh, C.; Jugurnauth-Little, S.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Barrowdale, D.; et al. Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease. Br. J. Cancer 2014, 110, 1663–1672. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Cook, L.S.; Goldoft, M.; Schwartz, S.M.; Weiss, N.S. Incidence of adenocarcinoma of the prostate in Asian immigrants to the United States and their descendants. J. Urol. 1999, 161, 152–155. [Google Scholar] [CrossRef]
- Peto, J. Cancer epidemiology in the last century and the next decade. Nature 2001, 411, 390–395. [Google Scholar] [CrossRef]
- Porter, C.M.; Shrestha, E.; Peiffer, L.B.; Sfanos, K.S. The microbiome in prostate inflammation and prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 345–354. [Google Scholar] [CrossRef]
- Fujita, K.; Hayashi, T.; Matsushita, M.; Uemura, M.; Nonomura, N. Obesity, Inflammation, and Prostate Cancer. J. Clin. Med. 2019, 8, 201. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of Diet and Nutrition on Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, S.; Fiorentino, M.; Andrén, O.; Fang, F.; Mucci, L.A.; Varenhorst, E.; Fall, K.; Rider, J.R. Inflammation, focal atrophic lesions, and prostatic intraepithelial neoplasia with respect to risk of lethal prostate cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Vral, A.; Magri, V.; Montanari, E.; Gazzano, G.; Gourvas, V.; Marras, E.; Perletti, G. Topographic and quantitative relationship between prostate inflammation, proliferative inflammatory atrophy and low-grade prostate intraepithelial neoplasia: A biopsy study in chronic prostatitis patients. Int. J. Oncol. 2012, 41, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef]
- Dennis, L.K.; Lynch, C.F.; Torner, J.C. Epidemiologic association between prostatitis and prostate cancer. Urology 2002, 60, 78–83. [Google Scholar] [CrossRef]
- Irani, J.; Goujon, J.M.; Ragni, E.; Peyrat, L.; Hubert, J.; Saint, F.; Mottet, N. High-grade inflammation in prostate cancer as a prognostic factor for biochemical recurrence after radical prostatectomy. Pathologist Multi Center Study Group. Urology 1999, 54, 467–472. [Google Scholar] [CrossRef]
- Roberts, R.O.; Bergstralh, E.J.; Bass, S.E.; Lieber, M.M.; Jacobsen, S.J. Prostatitis as a risk factor for prostate cancer. Epidemiology 2004, 15, 93–99. [Google Scholar] [CrossRef]
- MacLennan, G.T.; Eisenberg, R.; Fleshman, R.L.; Taylor, J.M.; Fu, P.; Resnick, M.I.; Gupta, S. The influence of chronic inflammation in prostatic carcinogenesis: A 5-year followup study. J. Urol. 2006, 176, 1012–1016. [Google Scholar] [CrossRef]
- Jiang, J.; Li, J.; Yunxia, Z.; Zhu, H.; Liu, J.; Pumill, C. The role of prostatitis in prostate cancer: Meta-analysis. PLoS ONE 2013, 8, e85179. [Google Scholar] [CrossRef] [PubMed]
- Gurel, B.; Lucia, M.S.; Thompson, I.M.; Goodman, P.J.; Tangen, C.M.; Kristal, A.R.; Parnes, H.L.; Hoque, A.; Lippman, S.M.; Sutcliffe, S.; et al. Chronic inflammation in benign prostate tissue is associated with high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2014, 23, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Hosomi, M.; Tanigawa, G.; Okumi, M.; Fushimi, H.; Yamaguchi, S. Prostatic inflammation detected in initial biopsy specimens and urinary pyuria are predictors of negative repeat prostate biopsy. J. Urol. 2011, 185, 1722–1727. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Imamura, R.; Tanigawa, G.; Nakagawa, M.; Hayashi, T.; Kishimoto, N.; Hosomi, M.; Yamaguchi, S. Low serum neutrophil count predicts a positive prostate biopsy. Prostate Cancer Prostatic Dis. 2012, 15, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Yli-Hemminki, T.H.; Laurila, M.; Auvinen, A.; Määttänen, L.; Huhtala, H.; Tammela, T.L.J.; Kujala, P.M. Histological inflammation and risk of subsequent prostate cancer among men with initially elevated serum prostate-specific antigen (PSA) concentration in the Finnish prostate cancer screening trial. BJU Int. 2013, 112, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.M.; Nickel, J.C.; Gerber, L.; Muller, R.L.; Andriole, G.L.; Castro-Santamaria, R.; Freedland, S.J. Baseline prostate inflammation is associated with a reduced risk of prostate cancer in men undergoing repeat prostate biopsy: Results from the REDUCE study. Cancer 2014, 120, 190–196. [Google Scholar] [CrossRef]
- Vasavada, S.R.; Dobbs, R.W.; Kajdacsy-Balla, A.A.; Abern, M.R.; Moreira, D.M. Inflammation on Prostate Needle Biopsy is Associated with Lower Prostate Cancer Risk: A Meta-Analysis. J. Urol. 2018, 199, 1174–1181. [Google Scholar] [CrossRef]
- Umbehr, M.H.; Gurel, B.; Murtola, T.J.; Sutcliffe, S.; Peskoe, S.B.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M.; Lippman, S.M.; Lucia, M.S.; et al. Intraprostatic inflammation is positively associated with serum PSA in men with PSA. Prostate Cancer Prostatic Dis. 2015, 18, 264–269. [Google Scholar] [CrossRef]
- Nakai, Y.; Nonomura, N. Inflammation and prostate carcinogenesis. Int. J. Urol. 2013, 20, 150–160. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Isaacs, W.B.; De Marzo, A.M. Infections and inflammation in prostate cancer. Am. J. Clin. Exp. Urol. 2013, 1, 3–11. [Google Scholar]
- Gandaglia, G.; Zaffuto, E.; Fossati, N.; Cucchiara, V.; Mirone, V.; Montorsi, F.; Briganti, A. The role of prostatic inflammation in the development and progression of benign and malignant diseases. Curr. Opin. Urol. 2017, 27, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Kulac, I.; Barber, J.R.; Drake, C.G.; Joshu, C.E.; Nelson, W.G.; Lucia, M.S.; Klein, E.A.; Lippman, S.M.; Parnes, H.L.; et al. A Prospective Study of Chronic Inflammation in Benign Prostate Tissue and Risk of Prostate Cancer: Linked PCPT and SELECT Cohorts. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Gao, X.; Li, X.; Qi, X.; Ma, M.; Qin, S.; Yu, H.; Sun, S.; Zhou, D.; Wang, W. Prognostic significance of neutrophil-to-lymphocyte ratio in prostate cancer: Evidence from 16,266 patients. Sci. Rep. 2016, 6, 22089. [Google Scholar] [CrossRef]
- Sciarra, A.; Gentilucci, A.; Salciccia, S.; Pierella, F.; Del Bianco, F.; Gentile, V.; Silvestri, I.; Cattarino, S. Prognostic value of inflammation in prostate cancer progression and response to therapeutic: A critical review. J. Inflamm. 2016, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Fujita, K.; Tanigawa, G.; Kawashima, A.; Nagahara, A.; Ujike, T.; Uemura, M.; Takao, T.; Yamaguchi, S.; Nonomura, N. Serum monocyte fraction of white blood cells is increased in patients with high Gleason score prostate cancer. Oncotarget 2017, 8, 35255–35261. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. Peripheral blood monocyte count reflecting tumor-infiltrating macrophages is a predictive factor of adverse pathology in radical prostatectomy specimens. Prostate 2017, 77, 1383–1388. [Google Scholar] [CrossRef]
- Ohno, Y. Role of systemic inflammatory response markers in urological malignancy. Int. J. Urol. 2019, 26, 31–47. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Matsushita, M.; Nonomura, N. Main Inflammatory Cells and Potentials of Anti-Inflammatory Agents in Prostate Cancer. Cancers 2019, 11, 1153. [Google Scholar] [CrossRef]
- Khandrika, L.; Kumar, B.; Koul, S.; Maroni, P.; Koul, H.K. Oxidative stress in prostate cancer. Cancer Lett. 2009, 282, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Rubin, M.A. Genomic rearrangements in prostate cancer. Curr. Opin. Urol. 2015, 25, 71–76. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Ewing, C.M.; Sokoll, L.J.; Elliott, D.J.; Cunningham, M.; De Marzo, A.M.; Isaacs, W.B.; Pavlovich, C.P. Cytokine profiling of prostatic fluid from cancerous prostate glands identifies cytokines associated with extent of tumor and inflammation. Prostate 2008, 68, 872–882. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Rani, A.; Dasgupta, P.; Murphy, J.J. Prostate Cancer: The Role of Inflammation and Chemokines. Am. J. Pathol. 2019, 189, 2119–2137. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef]
- Strasner, A.; Karin, M. Immune Infiltration and Prostate Cancer. Front. Oncol. 2015, 5, 128. [Google Scholar] [CrossRef]
- Taverna, G.; Pedretti, E.; Di Caro, G.; Borroni, E.M.; Marchesi, F.; Grizzi, F. Inflammation and prostate cancer: Friends or foe? Inflamm. Res. 2015, 64, 275–286. [Google Scholar] [CrossRef]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.O.; Poisson, A.O.; Delvoye, N.; Lapointe, R.; Mes-Masson, A.-M.; Saad, F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 2009, 348, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, N.; Takayama, H.; Nakayama, M.; Nakai, Y.; Kawashima, A.; Mukai, M.; Nagahara, A.; Aozasa, K.; Tsujimura, A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2011, 107, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Shimada, K.; Asai, O.; Tanaka, N.; Fujimoto, K.; Hirao, K.; Konishi, N. Immunohistochemical analysis of inflammatory cells in benign and precancerous lesions and carcinoma of the prostate. Pathobiology 2013, 80, 119–126. [Google Scholar] [CrossRef]
- Lanciotti, M.; Masieri, L.; Raspollini, M.R.; Minervini, A.; Mari, A.; Comito, G.; Giannoni, E.; Carini, M.; Chiarugi, P.; Serni, S. The role of M1 and M2 macrophages in prostate cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. Biomed Res. Int. 2014, 2014, 486798. [Google Scholar] [CrossRef]
- Hu, W.; Qian, Y.; Yu, F.; Liu, W.; Wu, Y.; Fang, X.; Hao, W. Alternatively activated macrophages are associated with metastasis and poor prognosis in prostate adenocarcinoma. Oncol. Lett. 2015, 10, 1390–1396. [Google Scholar] [CrossRef]
- Cao, J.; Liu, J.; Xu, R.; Zhu, X.; Zhao, X.; Qian, B.-Z. Prognostic role of tumour-associated macrophages and macrophage scavenger receptor 1 in prostate cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 83261–83269. [Google Scholar] [CrossRef]
- Erlandsson, A.; Carlsson, J.; Lundholm, M.; Fält, A.; Andersson, S.-O.; Andrén, O.; Davidsson, S. M2 macrophages and regulatory T cells in lethal prostate cancer. Prostate 2019, 79, 363–369. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Fang, L.-Y.; Izumi, K.; Lai, K.-P.; Liang, L.; Li, L.; Miyamoto, H.; Lin, W.-J.; Chang, C. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013, 73, 5633–5646. [Google Scholar] [CrossRef]
- Sousa, S.; Määttä, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Jones, J.D.; Purica, M.C.; Weidner, S.; Koh, A.J.; Kuo, R.; Wilkinson, J.E.; Wang, Y.; Daignault-Newton, S.; Pienta, K.J.; et al. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J. Clin. Investig. 2018, 128, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Sica, A.; Porta, C.; Morlacchi, S.; Banfi, S.; Strauss, L.; Rimoldi, M.; Totaro, M.G.; Riboldi, E. Origin and Functions of Tumor-Associated Myeloid Cells (TAMCs). Cancer Microenviron. 2012, 5, 133–149. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Plüddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Yoon, Y.-S.; Le Lay, J.; Kaestner, K.H.; Hedrick, S.; Montminy, M. CREB pathway links PGE2 signaling with macrophage polarization. Proc. Natl. Acad. Sci. USA 2015, 112, 15642–15647. [Google Scholar] [CrossRef] [PubMed]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e10. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- Sawa-Wejksza, K.; Kandefer-Szerszeń, M. Tumor-Associated Macrophages as Target for Antitumor Therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Greenberg, P.D.; Hingorani, S.R. Molecular pathways: Myeloid complicity in cancer. Clin. Cancer Res. 2014, 20, 5157–5170. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425, author reply 426. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Vuk-Pavlović, S.; Bulur, P.A.; Lin, Y.; Qin, R.; Szumlanski, C.L.; Zhao, X.; Dietz, A.B. Immunosuppressive CD14+HLA-DRlow/- monocytes in prostate cancer. Prostate 2010, 70, 443–455. [Google Scholar] [CrossRef]
- Brusa, D.; Simone, M.; Gontero, P.; Spadi, R.; Racca, P.; Micari, J.; Degiuli, M.; Carletto, S.; Tizzani, A.; Matera, L. Circulating immunosuppressive cells of prostate cancer patients before and after radical prostatectomy: Profile comparison. Int. J. Urol. 2013, 20, 971–978. [Google Scholar] [CrossRef]
- Chi, N.; Tan, Z.; Ma, K.; Bao, L.; Yun, Z. Increased circulating myeloid-derived suppressor cells correlate with cancer stages, interleukin-8 and -6 in prostate cancer. Int. J. Clin. Exp. Med. 2014, 7, 3181–3192. [Google Scholar]
- Idorn, M.; Køllgaard, T.; Kongsted, P.; Sengeløv, L.; Thor Straten, P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. 2014, 63, 1177–1187. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin. Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Huang, G.; Liu, S.; Wan, J.; Wang, X.; Zhu, Y.; Kaliney, W.; Zhang, C.; Cheng, L.; Wen, X.; et al. Polymorphonuclear MDSCs are enriched in the stroma and expanded in metastases of prostate cancer. J. Pathol. Clin. Res. 2020. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lei, Z.; Zhao, J.; Gong, W.; Liu, J.; Chen, Z.; Liu, Y.; Li, D.; Yuan, Y.; Zhang, G.-M.; et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007, 252, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Lavoie, S.; Michaud, M.; Gallini, C.A.; Kim, J.; Soucy, G.; Odze, R.; Glickman, J.N.; Garrett, W.S. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015, 12, 244–257. [Google Scholar] [CrossRef]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol. Cell. Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef]
- Lee, C.-R.; Lee, W.; Cho, S.K.; Park, S.-G. Characterization of Multiple Cytokine Combinations and TGF-β on Differentiation and Functions of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar] [CrossRef]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 2006, 176, 284–290. [Google Scholar] [CrossRef]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, T.; Shao, S.; Shi, B.; Zhao, Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1004983. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh, E.; Orangi, M.; Mohammadi, H.; Babaie, F.; Baradaran, B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J. Cell. Physiol. 2018, 233, 3024–3036. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.-J.; Salimzadeh, L.; Bagheri, N. Crosstalk between myeloid-derived suppressor cells and the immune system in prostate cancer: MDSCs and immune system in Prostate cancer. J. Leukoc. Biol. 2020, 107, 43–56. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Lerman, I.; Garcia-Hernandez, M.d.l.L.; Rangel-Moreno, J.; Chiriboga, L.; Pan, C.; Nastiuk, K.L.; Krolewski, J.J.; Sen, A.; Hammes, S.R. Infiltrating Myeloid Cells Exert Protumorigenic Actions via Neutrophil Elastase. Mol. Cancer Res. 2017, 15, 1138–1152. [Google Scholar] [CrossRef]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Ewing, C.M.; Isaacs, W.B.; Pavlovich, C.P. Immunomodulatory IL-18 binding protein is produced by prostate cancer cells and its levels in urine and serum correlate with tumor status. Int. J. Cancer 2011, 129, 424–432. [Google Scholar] [CrossRef]
- Sottnik, J.L.; Zhang, J.; Macoska, J.A.; Keller, E.T. The PCa Tumor Microenvironment. Cancer Microenviron. 2011, 4, 283–297. [Google Scholar] [CrossRef]
- Shiao, S.L.; Chu, G.C.-Y.; Chung, L.W.K. Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett. 2016, 380, 340–348. [Google Scholar] [CrossRef]
- Muthuswamy, R.; Corman, J.M.; Dahl, K.; Chatta, G.S.; Kalinski, P. Functional reprogramming of human prostate cancer to promote local attraction of effector CD8(+) T cells. Prostate 2016, 76, 1095–1105. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Miller, A.M.; Lundberg, K.; Ozenci, V.; Banham, A.H.; Hellström, M.; Egevad, L.; Pisa, P. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J. Immunol. 2006, 177, 7398–7405. [Google Scholar] [CrossRef]
- Maolake, A.; Izumi, K.; Shigehara, K.; Natsagdorj, A.; Iwamoto, H.; Kadomoto, S.; Takezawa, Y.; Machioka, K.; Narimoto, K.; Namiki, M.; et al. Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 2017, 8, 9739–9751. [Google Scholar] [CrossRef] [PubMed]
- Vickman, R.E.; Broman, M.M.; Lanman, N.A.; Franco, O.E.; Sudyanti, P.A.G.; Ni, Y.; Ji, Y.; Helfand, B.T.; Petkewicz, J.; Paterakos, M.C.; et al. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate 2020, 80, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Han, I.-H.; Song, H.-O.; Ryu, J.-S. IL-6 produced by prostate epithelial cells stimulated with Trichomonas vaginalis promotes proliferation of prostate cancer cells by inducing M2 polarization of THP-1-derived macrophages. PLoS Negl. Trop. Dis. 2020, 14, e0008126. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Lee, O.-Y.; Shon, S.Y.; Nam, O.; Ryu, P.M.; Seo, M.W.; Lee, D.-S. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. 2013, 15, R79. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Clements, V.K.; Hanson, E.M.; Sinha, P.; Ostrand-Rosenberg, S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J. Leukoc. Biol. 2009, 85, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Beury, D.W.; Parker, K.H.; Nyandjo, M.; Sinha, P.; Carter, K.A.; Ostrand-Rosenberg, S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J. Leukoc. Biol. 2014, 96, 1109–1118. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Bruno, T.C.; Maris, C.H.; Xu, L.; Thoburn, C.J.; DeMarzo, A.M.; Meeker, A.K.; Isaacs, W.B.; Drake, C.G. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin. Cancer Res. 2008, 14, 3254–3261. [Google Scholar] [CrossRef]
- Kiniwa, Y.; Miyahara, Y.; Wang, H.Y.; Peng, W.; Peng, G.; Wheeler, T.M.; Thompson, T.C.; Old, L.J.; Wang, R.-F. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin. Cancer Res. 2007, 13, 6947–6958. [Google Scholar] [CrossRef]
- Watanabe, M.; Kanao, K.; Suzuki, S.; Muramatsu, H.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Zennami, K.; et al. Increased infiltration of CCR4-positive regulatory T cells in prostate cancer tissue is associated with a poor prognosis. Prostate 2019, 79, 1658–1665. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, S.; Ge, D.; Cunningham, D.M.; Huang, F.; Ma, L.; Burris, T.P.; You, Z. Targeting Th17-IL-17 Pathway in Prevention of Micro-Invasive Prostate Cancer in a Mouse Model. Prostate 2017, 77, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, F.; Zhang, B.; Yan, P.; Rowan, B.G.; Abdel-Mageed, A.B.; Steele, C.; Jazwinski, S.M.; Moroz, K.; Norton, E.B.; et al. CD4+ T helper 17 cell response of aged mice promotes prostate cancer cell migration and invasion. Prostate 2020, 80, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef]
- Simons, B.W.; Durham, N.M.; Bruno, T.C.; Grosso, J.F.; Schaeffer, A.J.; Ross, A.E.; Hurley, P.J.; Berman, D.M.; Drake, C.G.; Thumbikat, P.; et al. A human prostatic bacterial isolate alters the prostatic microenvironment and accelerates prostate cancer progression. J. Pathol. 2015, 235, 478–489. [Google Scholar] [CrossRef]
- Jachetti, E.; Cancila, V.; Rigoni, A.; Bongiovanni, L.; Cappetti, B.; Belmonte, B.; Enriquez, C.; Casalini, P.; Ostano, P.; Frossi, B.; et al. Cross-Talk between Myeloid-Derived Suppressor Cells and Mast Cells Mediates Tumor-Specific Immunosuppression in Prostate Cancer. Cancer Immunol. Res. 2018, 6, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Nelson, W.G.; De Marzo, A.M. The dietary charred meat carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine acts as both a tumor initiator and promoter in the rat ventral prostate. Cancer Res. 2007, 67, 1378–1384. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Canene-Adams, K.; Hempel, H.; Yu, S.-H.; Simons, B.W.; Schaeffer, A.J.; Schaeffer, E.M.; Nelson, W.G.; De Marzo, A.M. Bacterial Prostatitis Enhances 2-Amino-1-Methyl-6-Phenylimidazo[4,5-b]Pyridine (PhIP)-Induced Cancer at Multiple Sites. Cancer Prev. Res. 2015, 8, 683–692. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Laurent, V.; Guérard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef]
- Divella, R.; De Luca, R.; Abbate, I.; Naglieri, E.; Daniele, A. Obesity and cancer: The role of adipose tissue and adipo-cytokines-induced chronic inflammation. J. Cancer 2016, 7, 2346–2359. [Google Scholar] [CrossRef]
- Corrêa, L.H.; Corrêa, R.; Farinasso, C.M.; de Sant’Ana Dourado, L.P.; Magalhães, K.G. Adipocytes and Macrophages Interplay in the Orchestration of Tumor Microenvironment: New Implications in Cancer Progression. Front. Immunol. 2017, 8, 1129. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Myeloid derived-suppressor cells: Their role in cancer and obesity. Curr. Opin. Immunol. 2018, 51, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Nassar, Z.D.; Aref, A.T.; Miladinovic, D.; Mah, C.Y.; Raj, G.V.; Hoy, A.J.; Butler, L.M. Peri-prostatic adipose tissue: The metabolic microenvironment of prostate cancer. BJU Int. 2018, 121, 9–21. [Google Scholar] [CrossRef]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Narita, S.; Nara, T.; Sato, H.; Koizumi, A.; Huang, M.; Inoue, T.; Habuchi, T. Research Evidence on High-Fat Diet-Induced Prostate Cancer Development and Progression. J. Clin. Med. 2019, 8, 597. [Google Scholar] [CrossRef]
- Ito, Y.; Ishiguro, H.; Kobayashi, N.; Hasumi, H.; Watanabe, M.; Yao, M.; Uemura, H. Adipocyte-derived monocyte chemotactic protein-1 (MCP-1) promotes prostate cancer progression through the induction of MMP-2 activity. Prostate 2015, 75, 1009–1019. [Google Scholar] [CrossRef]
- Huang, M.; Narita, S.; Numakura, K.; Tsuruta, H.; Saito, M.; Inoue, T.; Horikawa, Y.; Tsuchiya, N.; Habuchi, T. A high-fat diet enhances proliferation of prostate cancer cells and activates MCP-1/CCR2 signaling. Prostate 2012, 72, 1779–1788. [Google Scholar] [CrossRef]
- Xia, S.; Sha, H.; Yang, L.; Ji, Y.; Ostrand-Rosenberg, S.; Qi, L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J. Biol. Chem. 2011, 286, 23591–23599. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. High-Fat Diet-Induced Inflammation Accelerates Prostate Cancer Growth via IL6 Signaling. Clin. Cancer Res. 2018, 24, 4309–4318.


