Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claudia Salerno | + 2214 word(s) | 2214 | 2021-12-27 09:38:27 | | | |
| 2 | Rita Xu | Meta information modification | 2214 | 2022-01-10 05:00:26 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Salerno, C.; Cagetti, M.G. Rare Genetic Syndromes and Oral Anomalies. Encyclopedia. Available online: https://encyclopedia.pub/entry/17900 (accessed on 08 August 2026).
Salerno C, Cagetti MG. Rare Genetic Syndromes and Oral Anomalies. Encyclopedia. Available at: https://encyclopedia.pub/entry/17900. Accessed August 08, 2026.
Salerno, Claudia, Maria Grazia Cagetti. "Rare Genetic Syndromes and Oral Anomalies" Encyclopedia, https://encyclopedia.pub/entry/17900 (accessed August 08, 2026).
Salerno, C., & Cagetti, M.G. (2022, January 07). Rare Genetic Syndromes and Oral Anomalies. In Encyclopedia. https://encyclopedia.pub/entry/17900
Salerno, Claudia and Maria Grazia Cagetti. "Rare Genetic Syndromes and Oral Anomalies." Encyclopedia. Web. 07 January, 2022.
Copy Citation
Rare genetic syndromes, conditions with a global average prevalence of 40 cases/100,000 people, are associated with anatomical, physiological, and neurological anomalies that may affect different body districts, including the oral district.
rare genetic syndromes
dento-oro-maxillofacial anomalies
oral abnormalities
1. Introduction
A genetic syndrome is a condition caused by any abnormality in one’s genome that may be inherited or de novo. There is no clear and unique definition of rare genetic syndrome, but the meaning most commonly attributed to it in literature is that of a pathology with an average prevalence threshold ranging between five and 76 cases/100,000 people, with a global average prevalence of 40 cases/100,000 people. The Orphan Drug Act defines a rare disease or condition as a disease (a) which affects less than 200,000 people in the United States or (b) for which there is no reasonable expectation that the cost of developing a drug and making it available in the US will be recovered from its sale [1]. The member states of the European Union have adopted as a definition of rare disease, “the life-threatening or chronically debilitating conditions that affect maximum 5 persons over 10,000” [2].
Genetic syndromes are associated with anatomical, physiological, and neurological differences that may affect different body districts, including the mouth and its associated structures. Chromosomal localization is known for more than 50 of genetic diseases and responsible mutated genes have been discovered in more than 60 of these diseases with craniofacial and oral phenotypes alterations [3]. According to the London Dysmorphology Database [4], in 2011, approximately 900 out of 5000 genetic syndromes include dental-oro-maxillofacial anomalies in their clinical pictures, mostly among those affecting the ectodermal derivatives. Understanding dental genetics and developmental biology will allow the identification of the processes involved in the genesis of specific abnormalities [5][6][7].
Warning oral signals are dental and oral anomalies which, together with extraoral signals, can facilitate the early diagnosis of rare genetic syndromes. Dental anomalies consist in alterations of the external appearance, internal structure, or topography of one or more primary or permanent teeth, resulting from a disorder that can be genetically determined, congenital, or acquired. Dental anomalies can include reduced or increased teeth number, as well as variations in shape or volume, location or position, development, and structure of teeth (Table 1).
Table 1. Simplified table of dental anomalies and oral warning signs.
| Type of Anomaly | Clinical Signs | Anomalies Observed |
|---|---|---|
| Number anomalies | ||
| Reduced number | Anodontia | Absence of all dental elements |
| Oligodontia | Presence of less than half of the normal number of teeth | |
| Ipodontia | Presence of more than half of the normal number of teeth | |
| Increased number | Supplementary teeth | The tooth repeats the shape and function of the adjacent tooth |
| Supernumerary teeth | Elements are atypical, smaller and rudimentary. They are classified into: | |
| -Mesiodens: between the two upper central incisors | ||
| -Paramolar: in the molar region | ||
| -Distomolar: behind the third molar | ||
| Location anomalies | Ectopia | The element is located near the usual site, in a vestibular, lingual or palatal position |
| Transposition | Two contiguous teeth reciprocally invert their position | |
| Heterotopia | Element located far from the usual position. If the position is outside the oral cavity it is defined as migration. | |
| Position anomalies | Version | Inclination of a tooth towards the buccal or palatal side, forward (mesioversion) or backward (distortion) |
| Inversion | Inverted tooth position (root versus alveolar ridge and crown versus basal bone portion). It is constantly accompanied by inclusion. | |
| Rotation | The element rotates along its longitudinal axis | |
| Intrusion | The tooth crown is on a lower plane than the occlusal plane | |
| Extrusion | The coronal margin is located higher than the occlusal plane | |
| Volume anomalies | Gigantism | Macrodontia |
| Dwarfism | Microdontia | |
| Taurodontism | Tooth with wide crown, short roots, extended pulp chamber | |
| Shape anomalies | Crown anomalies | Accessory cusps, conical or tuberculate shape of the crown |
| Roots anomalies | Anomalies in number, shape and size | |
| Endodontic anomalies | Anomalies in number, shape and size of the root canal morphology | |
| Developmental anomalies | Enamel pearl | Small hard rounded excrescence, located near the enamel-cement junction. |
| Fusion | Fusion of two teeth. Coalescence anomaly: due to close contact between germs; it can affect crowns and roots or only the crowns | |
| Concrescence | Two teeth are joined along the root surfaces by cementum. Coalescence anomaly | |
| Invaginated tooth | It is due to an infolding of enamel into dentine. There are two forms, coronal and radicular, with the coronal one being more common. | |
| Structural anomalies | Enamel anomalies | Quantitative and/or qualitative enamel defect affecting some or all teeth. Amelogenesis imperfecta: abnormal formation of the enamel, unrelated to any systemic or generalized conditions. Autosomal dominant or autosomal recessive or x-linked pattern. It affects both deciduous and permanent. |
| Dentin anomalies | Dentinogenesis imperfecta: brown teeth, crowns shrunk due to enamel flaking not supported by intact dentin. Autosomal dominant. It affects both deciduous and permanent. |
Developmental dental defects can result from teratogenic actions during odontogenesis as well as genetic disorders, isolated or associated to other signs in a syndrome [8]. Dental malformations may be the most evident manifestations, and therefore first diagnosed, while other signs affecting different organs will only manifest themselves later. These anomalies have both functional and aesthetical implications [9].
Dentists should be aware of the existence of rare genetic syndromes and should know how to clinically recognize, manage, and treat the dento-oro-craniofacial anomalies they might include. Dentists, in fact, could be asked to cooperate with other specialists in the diagnosis of these diseases. Rare genetic syndromes, despite their generally chronic and progressive nature, have long-term complications that can be reduced or delayed if they are early diagnosed and managed [10]. An oral rehabilitation is frequently necessary to guarantee mastication, swallowing, phonetics, and mechanical ventilation. Moreover, good dental appearance can positively contribute to the integration of affected subjects in society as well in school [11].
The main sources of information for clinicians on management recommendations for patients with rare genetic syndromes are very limited and consist of: Internet sites such as Orpha.net [12], a portal for rare diseases and orphan drugs, which offers a list of the diseases; databases such as Phenodent, where it is possible to find help in the diagnosis and management of dental anomalies [13]; the Genetic and rare diseases information center (GARD) [14], a program of the National Center for Advancing Translational Sciences, where the rare diseases are classified and explained; and the Online Mendelian Inheritance in Man (OMIM) [15][16], that since 1966 has continuously provided an updated catalog of human genes and genetic disorders and traits, and it has been online since 1987. Orpha.net provides an exhaustive list of all rare diseases, updated every year.
Rare diseases present on Orpha.net are defined according to two criteria:
-
Homogeneity: Each disease is defined by its clinical homogeneity, regardless of the etiology or the number of identified responsible genes.
-
Rarity: Each disease is defined based on the European legislation, which establishes a prevalence threshold not exceeding 5 affected persons per 10,000 people.
Rare diseases need to be described in the international scientific literature (peer-reviewed articles) with at least two cases, confirming the non-random association of clinical signs [17].
Regardless of the oral malformation observed, therapeutic management is often long and complex. It starts at the time of diagnosis and continues during the child’s growth until adulthood, with the involvement of a multidisciplinary team.
Five syndromes are described below via three different cases, namely the Crouzon Syndrome, a Congenital Nemaline Myopathy, the CHARGE Syndrome.
2. Crouzon Syndrome
Twenty-eight papers describing the relationship between the Crouzon syndrome and oral anomalies were found in the PubMed search.
The Crouzon syndrome (CS) is an autosomal dominant genetic disease caused by mutations in one of the FGFR (fibroblast growth factor) genes, especially the FGFR2, which is responsible for the early closure of the cranial sutures. It has an incidence of 1 case in 25,000 births [18].
This syndrome has an extremely variable phenotypic manifestation in both the cranial and facial features [19][20]. On the one hand, some patients present a mild form of this syndrome that can guarantee a fully functional lifestyle. On the other hand, other patients suffer from a severe form of the disease that can cause a significant impact on their quality of life [20].
Clinical manifestations include tall and flattened forehead, maxillary hypoplasia and mandibular prognathism. The arrested growth of the upper maxilla causes bilateral severe proptosis and corneal exposure, thus resulting in shallow orbits. Hypertelorism, exorbitism, strabismus, and amblyopia are frequently found as well [21][22][23][24]. Alongside the ocular problems, these anatomical anomalies can lead to other functional problems like increased intracranial pressure and upper airway obstruction [25].
C. M. is a 10-year old male, patient of the Dental Clinic of San Paolo Hospital, Milan. During the extra-oral examination, the boy presented the typical signs of Crouzon Syndrome: a high and large forehead with a convexity in the region of the anterior fontanelle, flattening of the occipital region, bicoronal craniosynostosis and hypertelorism. He did not present maxillary hypoplasia or exorbitism. No mental retardation was observed.
The oral manifestations included the maxillary dental arch in V shape (Figure 1d), dental crowding, class II maloocclusion with an anterior severe open bite and lower labial interposition at rest (Figure 1a–c).
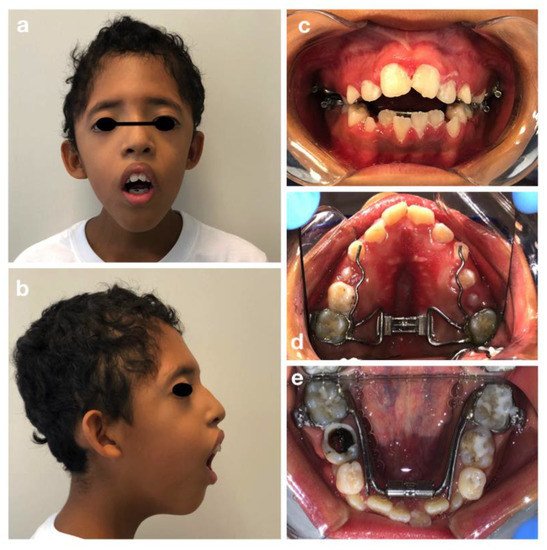
Figure 1. Crouzon syndrome: extra-oral (a,b) and intra-oral pictures (c–e). Upper and lower arch expanders are present (d,e). An arrested cavitated caries lesion in a second primary molar (8.5) is evident (e).
The intraoral examination showed an intermediate mixed dentition phase, decayed and filled teeth and gingivitis due to the presence of bacterial plaque, dental calculus, and oral breath (Figure 2).
Since the patient started an orthodontic treatment with upper and lower arch expanders in another dental clinic, the pre-treatment radiographs and pictures are not available.
3. Congenital Nemaline Myopathies
Only one paper describing the relationship between congenital nemaline myopathies and oral anomalies was found in the PubMed search.
Congenital nemaline myopathies (CNM) are a heterogenous group of congenital myopathies caused by inherited mutations in more than twelve genes. CNM incidence is estimated to be one in 50,000 live births. The genes which encode skeletal α-actin (ACTA1) and nebulin (NEB) are the most frequently mutated [26].
The main manifestations of the disease include muscle weakness and hypotonia, with the presence of nemaline bodies in muscle fibers. CNM can manifest itself with multiple clinical phenotypes: in the most severe cases it can lead to neonatal death, while in the milder ones the disease causes only a slight impairment of motor function. When the respiratory muscles are involved as well, patients are ventilator-dependent [27]. No therapeutic treatment is available yet [26]. The clinical manifestations of CNM include severe muscle weakness, hypoventilation, swallowing dysfunction, and no speech ability. Oral or dental anomalies in patients with this disorder are not yet described in literature, except for a reduced development of the maxilla and mandible, due to muscular hypotonia [28].
L.R. is a three-year-old boy affected by Congenital Nemaline Myopathy, who required a visit at the Dental Clinic of S. Paolo Hospital, Milan for severe mobility of all primary incisors.
The oral examination showed a severe reduced development of the maxilla, with a severe high-arched palate and reduced mouth opening (Figure 2).
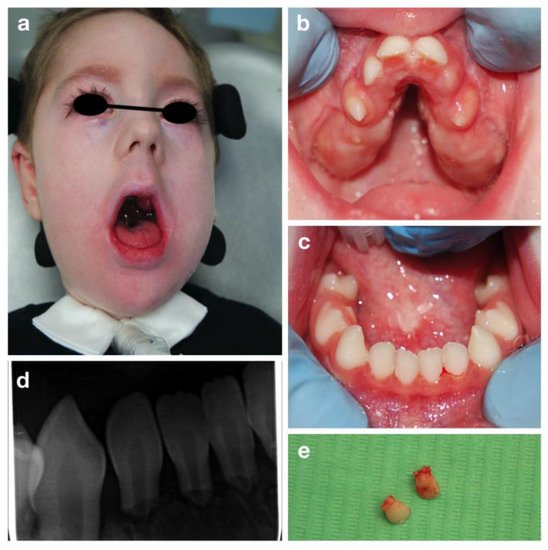
Figure 2. Congenital nemaline myopathies: extra-oral (a), intra-oral pictures (b,c), periapical radiograph of lower incisors (d) and the upper incisors after extraction (e).
Mobility of all the primary incisors was found, together with a delayed eruption of primary molars and canines. Due to the patient’s condition an orthopantomography was impossible to be carried out. Therefore, a periapical radiography was performed despite the difficulty in positioning the intraoral plate because of the reduced arches width (Figure 2b,c). A severe root resorption of unclear etiology and no evidence of permanent successors was noted (Figure 2).
4. CHARGE Syndrome
Twenty-one papers describing the relationship between the CHARGE syndrome and oral anomalies were found in the PubMed search.
The CHARGE syndrome is an autosomal dominant genetic disorder with multiple dysmorphic and congenital anomalies. It is a highly variable syndrome, usually diagnosed during the prenatal or neonatal period [29][30]. The actual incidence remains unknown, but it is believed to occur approximately in one case in 10,000–15,000 live births [31].
The term ‘CHARGE’ is an acronym generated by the initials of the most frequent clinical features: Coloboma, Heart defects, choanal Atresia, Retardation (of growth and/or development), Genitourinary malformation and Ear abnormalities [32]. Thus, the diagnosis is predominantly clinical, using the criteria proposed by Blake et al. [33], modified by Verloes [34], and then confirmed by genetic testing [31].
Clinical findings include orofacial cleft, distinctive facial appearance, tracheoesophageal fistula, limb abnormalities, cranial nerve dysfunction, semicircular canal hypoplasia, delayed attainment of motor milestones, genital hypoplasia, and rarely, immune deficiencies [29][30].
A.T. is an eight-year old girl who attended the Dental Clinic of San Paolo Hospital, Milano for dental treatment. She was diagnosed both clinically (all the main signs of the CHARGE syndrome were present) and genetically, since the CHD7 gene mutation was confirmed. The girl presented bilateral chorio-retinal coloboma, cerebral-cerebellar malformation, middle and inner ear malformation, reduced weight and stature growth, globally delayed psychomotor development with absence of language. Cleft lip and cleft palate were also reported, surgically corrected in 2013. Intraoral examination showed a good oral hygiene and no dental caries lesions (Figure 3).
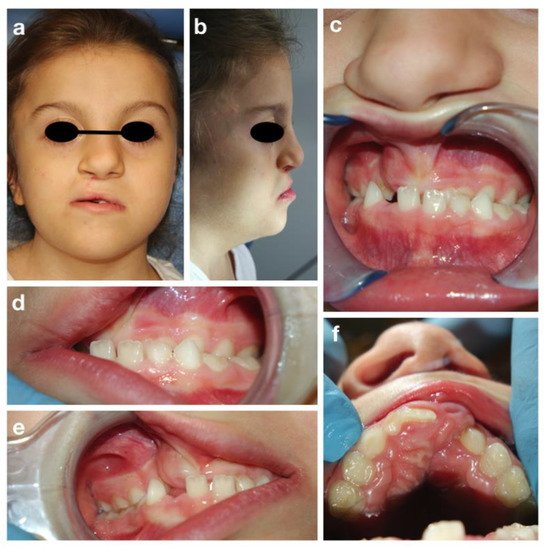
Figure 3. CHARGE syndrome: extra-oral (a,b) and intra-oral pictures (c–f).
The intraoral examination showed a mixed dentition with lacking space in both arches for the eruption of the permanent incisors and a class III malocclusion with anterior crossbite (Figure 3c), due to maxillary hypoplasia.
An attempt to perform an orthopantomography was made, but the result was poor, and it was not possible to evaluate the presence of all permanent teeth. However, a prediction of dental crowding was detected.
References
- Richter, T.; Nestler-Parr, S.; Babela, R.; Khan, Z.M.; Tesoro, T.; Molsen, E.; Hughes, D. Rare Disease Terminology and Definitions—A Systematic Global Review: Report of the ISPOR Rare Disease Special Interest Group. Value Health 2015, 18, 906–914.
- European Project for Rare Diseases National Plans Management EUROPLAN. Available online: www.euro-planproject.eu/Content?folder=1 (accessed on 15 November 2021).
- Thesleff, I. The genetic basis of normal and abnormal craniofacial development. Acta Odontol. Scand. 1998, 56, 321–325.
- London Dysmorphology Database. Available online: http://www.lmdatabases.com/ (accessed on 15 July 2021).
- London, J.; Birkedal-Hansen, H. Opportunities in dental, oral, and craniofacial research. Compend. Contin. Educ. Dent. 2000, 21, 760–762, 764, 766.
- Slavkin, H. Craniofacial-Oral-Dental Research in the 21st Century. J. Dent. Res. 1997, 76, 628–630.
- Slavkin, H.C. Molecular Biology Experimental Strategies for Craniofacial-Oral-Dental Dysmorphology. Connect. Tissue Res. 1995, 32, 233–239.
- Bloch-Zupan, A.; Sedano, H.; Scully, C. Dento/Oro/Craniofacial Anomalies and Genetics, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 1–8.
- Alliot-Licht, B.; Lusson, C.; Hyon, I.; Dajean-Trutaud, S.; Le Caignec, C.; Lopez-Cazaux, S. Signes extra-oraux à rechercher face à des signes bucco-dentaires d’alerte de maladies d’origine génétique . Comptes Rendus Biol. 2015, 338, 48–57.
- Boycott, K.M.; Rath, A.; Chong, J.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; Dunnen, J.T.D.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705.
- American Academy of Pediatric Dentistry. Guideline on dental management of heritable dental developmental anomalies. Pediatr. Dent. 2013, 35, E179–E184.
- Orpha.net. Available online: https://www.orpha.net/ (accessed on 17 July 2021).
- Phenodent. Available online: http://www.phenodent.org/ (accessed on 17 July 2021).
- GARD. Available online: https://rarediseases.info.nih.gov/ (accessed on 17 July 2021).
- Online Mendelian Inheritance in Man. Available online: https://omim.org/ (accessed on 11 November 2020).
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798.
- Regulation (EC) n° 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Drugs. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32000R0141 (accessed on 15 November 2020).
- Kaushik, A.; Bhatia, H.; Sharma, N. Crouzon’s Syndrome: A Rare Genetic Disorder. Int. J. Clin. Pediatr. Dent. 2016, 9, 384–387.
- Carinci, F.; Pezzetti, F.; Locci, P.; Becchetti, E.; Carls, F.; Avantaggiato, A.; Becchetti, A.; Carinci, P.; Baroni, T.; Bodo, M. Apert and Crouzon Syndromes: Clinical Findings, Genes and Extracellular Matrix. J. Craniofac. Surg. 2005, 16, 361–368.
- Helman, S.N.; Badhey, A.; Kadakia, S.; Myers, E. Revisiting Crouzon syndrome: Reviewing the background and management of a multifaceted disease. Oral Maxillofac. Surg. 2014, 18, 373–379.
- Khandelwal, R.; Agrawal, P.; Majumdar, M.R. Crouzon syndrome. BMJ Case Rep. 2012.
- Crouzon, O. Dysostose Cranio-Faciale Héréditaire; Bulletins et mémoires de la Société Médicale des Hôpitaux: Paris, France, 1912; p. 545.
- Renier, D.; Lajeunie, E.; Arnaud, E.; Marchac, D. Management of craniosynostoses. Child’s Nerv. Syst. 2000, 16, 645–658.
- Khominsky, A.; Yong, R.; Ranjitkar, S.; Townsend, G.; Anderson, P.J. Extensive phenotyping of the orofacial and dental complex in Crouzon syndrome. Arch. Oral Biol. 2018, 86, 123–130.
- Staal, F.C.; Ponniah, A.J.; Angullia, F.; Ruff, C.; Koudstaal, M.; Dunaway, D. Describing Crouzon and Pfeiffer syndrome based on principal component analysis. J. Cranio-Maxillofac. Surg. 2015, 43, 528–536.
- Claeys, K.G. Congenital myopathies: An update. Dev. Med. Child Neurol. 2020, 62, 297–302.
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126.
- Anderson, P.J.; Barker, J.H.; David, D.J. Management of facial dysmorphogenesis in nemaline myopathy: A case re-port. World J. Orthod. 2005, 6, 156–160.
- Bergman, J.; Janssen, N.; Hoefsloot, L.H.; Jongmans, M.C.; Hofstra, R.; Van Ravenswaaij-Arts, C.M.A. CHD7 mutations and CHARGE syndrome: The clinical implications of an expanding phenotype. J. Med Genet. 2011, 48, 334–342.
- Usman, N.; Sur, M. CHARGE Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Hudson, A.; Trider, C.-L.; Blake, K. CHARGE Syndrome. Pediatr. Rev. 2017, 38, 56–59.
- Hsu, P.; Ma, A.; Wilson, M.; Williams, G.; Curotta, J.; Munns, C.F.; Mehr, S. CHARGE syndrome: A review. J. Paediatr. Child Health 2014, 50, 504–511.
- Blake, K.D.; Davenport, S.L.H.; Hall, B.D.; Hefner, M.A.; Pagon, R.A.; Williams, M.S.; Lin, A.E.; Graham, J.M., Jr. CHARGE Association: An Update and Review for the Primary Pediatrician. Clin. Pediatr. 1998, 37, 159–173.
- Sanlaville, D.; Verloes, A. CHARGE syndrome: An update. Eur. J. Hum. Genet. 2007, 15, 389–399.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
10 Jan 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No