 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marios Sagris | + 3095 word(s) | 3095 | 2021-12-27 09:38:52 | | | |
| 2 | Jessie Wu | -1 word(s) | 3094 | 2022-01-10 02:27:11 | | |
Video Upload Options
Atrial fibrillation (AF) is the most frequent arrhythmia managed in clinical practice, and it is linked to an increased risk of death, stroke, and peripheral embolism. The Global Burden of Disease shows that the estimated prevalence of AF is up to 33.5 million patients. So far, successful therapeutic techniques have been implemented, with high healthcare cost burdening. As a result, identifying modifiable risk factors for AF and suitable preventive measures may play a significant role in enhancing community health and lowering health-care system expenditures. Several mechanisms, including electrical and structural remodeling of atrial tissue, have been proposed to contribute to the development of AF. This entry discusses the predisposing factors in AF including the different pathogenic mechanisms, sedentary lifestyle, dietary habits as well as the potential genetic burdening.
1. Backgroud
Over the past hundred years, atrial fibrillation (AF) is the arrhythmia that has been studied the most among all other heart rhythm disorders, leading to valuable conclusions [1]. The prevalence of AF ranges from 2% in the general population to 10–12% in those aged 80 and older [2]. It is the most common arrhythmia in humans, and incidence increases with advancing age [2]. According to the Global Burden of Disease, the estimated prevalence of AF is up to 33.5 million individuals, as it affects 2.5–3.5% of populations in several countries [3]. Atrial fibrosis has emerged as a significant pathophysiological component, with links to AF recurrences, resistance to medication, and complications [3].
2. Fibrosis
-
Interstitial fibrosis can be sub-classified into:
- (a)
- (b)
-
Infiltrative interstitial fibrosis, which refers to the deposition of glycosphingolipids or insoluble proteins in the interstitial space, as seen in amyloidosis or Fabry disease respectively [7].
2.1. Cellular Mediators of Atrial Fibrosis
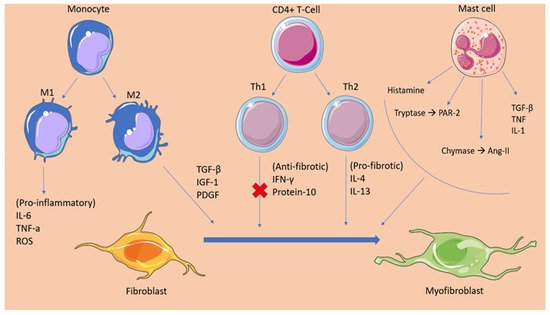
2.2. Fibrotic Mechanisms Inducing Atrial Fibrillation
3. Oxidative Stress
4. Inflammation
References
- Lau, D.H.; Linz, D.; Sanders, P. New Findings in Atrial Fibrillation Mechanisms. Card Electrophysiol. Clin. 2019, 11, 563–571.
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.J.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517.
- Morin, D.P.; Bernard, M.L.; Madias, C.; Rogers, P.A.; Thihalolipavan, S.; Estes, N.A., 3rd. The State of the Art: Atrial Fibrillation Epidemiology, Prevention, and Treatment. Mayo Clin. Proc. 2016, 91, 1778–1810.
- Spencer, T.M.; Blumenstein, R.F.; Pryse, K.M.; Lee, S.-L.; Glaubke, D.A.; Carlson, B.E.; Elson, E.L.; Genin, G.M. Fibroblasts Slow Conduction Velocity in a Reconstituted Tissue Model of Fibrotic Cardiomyopathy. ACS Biomater. Sci. Eng. 2017, 3, 3022–3028.
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435.
- Burstein, B.; Nattel, S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809.
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82.
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.D.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242.
- Nattel, S. Electrical coupling between cardiomyocytes and fibroblasts: Experimental testing of a challenging and important concept. Cardiovasc. Res. 2018, 114, 349–352.
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell 2012, 23, 705–715.
- Hoyles, R.K.; Derrett-Smith, E.C.; Khan, K.; Shiwen, X.; Howat, S.L.; Wells, A.U.; Abraham, D.J.; Denton, C.P. An Essential Role for Resident Fibroblasts in Experimental Lung Fibrosis Is Defined by Lineage-Specific Deletion of High-Affinity Type II Transforming Growth Factor β Receptor. Am. J. Respir. Crit. Care Med. 2011, 183, 249–261.
- Leask, A. Potential Therapeutic Targets for Cardiac Fibrosis. Circ. Res. 2010, 106, 1675–1680.
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell Cardiol. 2014, 70, 9–18.
- Lu, H.; Tian, A.; Wu, J.; Yang, C.; Xing, R.; Jia, P.; Yang, L.; Zhang, Y.; Zheng, X.; Li, Z. Danshensu Inhibits β-Adrenergic Receptors-Mediated Cardiac Fibrosis by ROS/p38 MAPK Axis. Biol. Pharm. Bull. 2014, 37, 961–967.
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis. Circ. Res. 2020, 127, 427–447.
- Pellman, J.; Zhang, J.; Sheikh, F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: Mechanisms and model systems. J. Mol. Cell Cardiol. 2016, 94, 22–31.
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int. J. Mol. Sci. 2021, 22, 1170.
- Kim, P.; Chu, N.; Davis, J.; Kim, D.H. Mechanoregulation of Myofibroblast Fate and Cardiac Fibrosis. Adv. Biosyst 2018, 2.
- Zaidi, Y.; Aguilar, E.G.; Troncoso, M.; Ilatovskaya, D.V.; DeLeon-Pennell, K.Y. Immune regulation of cardiac fibrosis post myocardial infarction. Cell Signal. 2021, 77, 109837.
- Shiota, N.; Jin, D.; Takai, S.; Kawamura, T.; Koyama, M.; Nakamura, N.; Miyazaki, M. Chymase is activated in the hamster heart following ventricular fibrosis during the chronic stage of hypertension. FEBS Lett. 1997, 406, 301–304.
- Ahmad, S.; Varagic, J.; Westwood, B.M.; Chappell, M.C.; Ferrario, C.M. Uptake and Metabolism of the Novel Peptide Angiotensin-(1-12) by Neonatal Cardiac Myocytes. PLoS ONE 2011, 6, e15759.
- Balcells, E.; Meng, Q.C.; Walter, H.; Johnson, J.; Oparil, S.; Dell’Italia, L.J. Angiotensin II formation from ACE and chymase in human and animal hearts: Methods and species considerations. Am. J. Physiol. Heart Circ. Physiol. 1997, 273, H1769–H1774.
- Shimizu, M.; Tanaka, R.; Fukuyama, T.; Aoki, R.; Orito, K.; Yamane, Y. Cardiac Remodeling and Angiotensin II-Forming Enzyme Activity of the Left Ventricle in Hamsters with Chronic Pressure Overload Induced by Ascending Aortic Stenosis. J. Vet. Med Sci. 2006, 68, 271–276.
- McLarty, J.L.; Meléndez, G.C.; Brower, G.L.; Janicki, J.S.; Levick, S.P. Tryptase/Protease-Activated Receptor 2 Interactions Induce Selective Mitogen-Activated Protein Kinase Signaling and Collagen Synthesis by Cardiac Fibroblasts. Hypertension 2011, 58, 264–270.
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448.
- Morgan, L.G.; Levick, S.P.; Voloshenyuk, T.G.; Murray, D.B.; Forman, M.F.; Brower, G.L.; Janicki, J.S. A novel technique for isolating functional mast cells from the heart. Inflamm Res. 2008, 57, 241–246.
- Nattel, S. How does fibrosis promote atrial fibrillation persistence: In silico findings, clinical observations, and experimental data. Cardiovasc. Res. 2016, 110, 295–297.
- Allessie, M.A.; de Groot, N.M.; Houben, R.P.; Schotten, U.; Boersma, E.; Smeets, J.L.; Crijns, H.J. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: Longitudinal dissociation. Circ. Arrhythm Electrophysiol. 2010, 3, 606–615.
- Krul, S.P.; Berger, W.R.; Smit, N.W.; van Amersfoorth, S.C.; Driessen, A.H.; van Boven, W.J.; Fiolet, J.W.; van Ginneken, A.C.; van der Wal, A.C.; de Bakker, J.M.; et al. Atrial fibrosis and conduction slowing in the left atrial appendage of patients undergoing thoracoscopic surgical pulmonary vein isolation for atrial fibrillation. Circ. Arrhythm Electrophysiol. 2015, 8, 288–295.
- Hansen, B.J.; Zhao, J.; Csepe, T.A.; Moore, B.T.; Li, N.; Jayne, L.A.; Kalyanasundaram, A.; Lim, P.; Bratasz, A.; Powell, K.A.; et al. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur. Heart J. 2015, 36, 2390–2401.
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm. 2017, 14, 1849–1855.
- Sovari, A.A.; Dudley, S.C., Jr. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physiol. 2012, 3, 311.
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.-X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427.
- Yoo, S.; Aistrup, G.; Shiferaw, Y.; Ng, J.; Mohler, P.J.; Hund, T.J.; Waugh, T.; Browne, S.; Gussak, G.; Gilani, M.; et al. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 2018, 3.
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717.
- Sagris, M.; Antonopoulos, A.S.; Theofilis, P.; Oikonomou, E.; Siasos, G.; Tsalamandris, S.; Antoniades, C.; Brilakis, E.S.; Kaski, J.C.; Tousoulis, D. Risk factors profile of young and older patients with Myocardial Infarction. Cardiovasc. Res. 2021.
- Babusikova, E.; Kaplan, P.; Lehotsky, J.; Jesenak, M.; Dobrota, D. Oxidative modification of rat cardiac mitochondrial membranes and myofibrils by hydroxyl radicals. Gen. Physiol. Biophys. 2004, 23, 327–335.
- Conen, D.; Ridker, P.M.; Everett, B.M.; Tedrow, U.B.; Rose, L.; Cook, N.R.; Buring, J.E.; Albert, C.M. A multimarker approach to assess the influence of inflammation on the incidence of atrial fibrillation in women. Eur. Heart J. 2010, 31, 1730–1736.
- Kallergis, E.M.; Manios, E.G.; Kanoupakis, E.M.; Mavrakis, H.E.; Kolyvaki, S.G.; Lyrarakis, G.M.; Chlouverakis, G.I.; Vardas, P.E. The role of the post-cardioversion time course of hs-CRP levels in clarifying the relationship between inflammation and persistence of atrial fibrillation. Heart 2008, 94, 200–204.
- Rotter, M.; Jaïs, P.; Vergnes, M.-C.; Nurden, P.; Takahashi, Y.; Sanders, P.; Rostock, T.; Hocini, M.; Sacher, F.; Haïssaguerre, M. Decline in C-Reactive Protein After Successful Ablation of Long-Lasting Persistent Atrial Fibrillation. J. Am. Coll. Cardiol. 2006, 47, 1231–1233.
- Mouselimis, D.; Tsarouchas, A.S.; Pagourelias, E.D.; Bakogiannis, C.; Theofilogiannakos, E.K.; Loutradis, C.; Fragakis, N.; Vassilikos, V.P.; Papadopoulos, C.E. Left atrial strain, intervendor variability, and atrial fibrillation recurrence after catheter ablation: A systematic review and meta-analysis. Hellenic J. Cardiol. 2020, 61, 154–164.
- Gao, G.; Dudley, S.C., Jr. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid Redox Signal. 2009, 11, 2265–2277.
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781.
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a Regulator of the Renin-Angiotensin System and Blood Pressure. Curr. Hypertens. Rep. 2018, 20, 100.
- Liang, F.; Wang, Y. Coronary heart disease and atrial fibrillation: A vicious cycle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1–H12.
- Aronson, D.; Boulos, M.; Suleiman, A.; Bidoosi, S.; Agmon, Y.; Kapeliovich, M.; Beyar, R.; Markiewicz, W.; Hammerman, H.; Suleiman, M. Relation of C-reactive protein and new-onset atrial fibrillation in patients with acute myocardial infarction. Am. J. Cardiol. 2007, 100, 753–757.
- Marcus, G.M.; Whooley, M.A.; Glidden, D.V.; Pawlikowska, L.; Zaroff, J.G.; Olgin, J.E. Interleukin-6 and atrial fibrillation in patients with coronary artery disease: Data from the Heart and Soul Study. Am. Heart J. 2008, 155, 303–309.
- Zhang, P.; Shao, L.; Ma, J. Toll-Like Receptors 2 and 4 Predict New-Onset Atrial Fibrillation in Acute Myocardial Infarction Patients. Int. Heart J. 2018, 59, 64–70.
- Xu, Y.; Sharma, D.; Du, F.; Liu, Y. The role of Toll-like receptor 2 and hypoxia-induced transcription factor-1α in the atrial structural remodeling of non-valvular atrial fibrillation. Int. J. Cardiol. 2013, 168, 2940–2941.
- Maehama, T.; Okura, H.; Imai, K.; Saito, K.; Yamada, R.; Koyama, T.; Hayashida, A.; Neishi, Y.; Kawamoto, T.; Yoshida, K. Systemic inflammation and left atrial thrombus in patients with non-rheumatic atrial fibrillation. J. Cardiol. 2010, 56, 118–124.
- Kaski, J.C.; Arrebola-Moreno, A.L. Inflamación y trombosis en la fibrilación auricular. Rev. Esp. Cardiol. 2011, 64, 551–553.
- Shantsila, E.; Lip, G.Y. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb. Haemost. 2009, 102, 916–924.
- Nair, G.M.; Nery, P.B.; Redpath, C.J.; Birnie, D.H. The Role Of Renin Angiotensin System In Atrial Fibrillation. J. Atr. Fibrillation 2014, 6, 972.
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, C.; Oikonomou, E.; Antoniades, C.; Crea, F.; Kaski, J.C.; Tousoulis, D. Inflammatory Mechanisms in COVID-19 and Atherosclerosis: Current Pharmaceutical Perspectives. Int. J. Mol. Sci. 2021, 22, 6607.

