Over recent years, oxidative stress has been investigated as a potential essential mechanism in the development of AF. Reactive oxygen species (ROS) constitute the normal byproducts generated through the metabolism of oxygen. These molecules have been proven to have a multifaceted effect on the cells present in the heart tissue. Tahhan et al. recently revealed that the prevalence and incidence of AF were related to the redox potentials of glutathione (E
hGSH) and cysteine, markers of oxidative stress. The study concluded that the prevalence of AF was 30% higher for each 10% increase in E
hGSH, while the same alteration resulted in a 40% increase in the risk of incident AF
[31][36]. The molecular processes underpinning atrial fibrillation development have been the subject of multiple clinical studies. Research evidence suggest that excessive ROS can directly affect ion channels and the propagation of action potential
[32][37]. Hydrogen peroxide provokes trigger activity through the enhancement of late Na+ current, inducing early afterdepolarization (EAD) and delayed afterdepolarization (DAD). Moreover, ROS can induce a downregulation of the total Na+ current, an event that promotes the formation of reentry circuits. It is also worth mentioning that ROS can directly upregulate the L-type Ca
2+ current and promote EADs by altering the intracellular calcium balance
[32][37]. Recent experimental evidence suggests that the oxidation of ryanodine receptor 2 (RYR2) induces the intracellular release of Ca
2+ from the sarcoplasmic reticulum, promoting the establishment of atrial fibrillation
[33][38]. The generation of ROS in the myocardium has been attributed to many enzymatic sources. Among them, NADPH oxidase (NOX) has proven to have a critical role in the progress of AF. In studies performed in animal models, superoxide and H
20
2 produced from activated NOX2 and NOX4 isoforms lead to myocyte apoptosis, fibrosis, and inflammation, which further promote atrial fibrillation perpetuation. One proposed mechanism through which ROS could exert their pro-arrhythmic function is by the oxidation of calmodulin-dependent protein kinase II (CaMKII)
[34][39]. Oxidized CaMKII mediate the phosphorylation of the RYR2, leading to calcium overload and the formation of multiple wavelets triggering atrial fibrillation emergence
[35][40]. In addition to the electrical remodeling stimulated by the mechanisms described, ROS have also been demonstrated to contribute to atria structural remodeling. Researchers from Slovakia showed that hydroxyl radicals can alter the myofibrillar protein structure and function, promoting myocardial injury and further contributing to the formation of a fertile substrate for the development of arrhythmias
[36][37][5,41].
4. Inflammation
Inflammation has been linked to the onset and maintenance of atrial fibrillation, according to accumulating evidence. Inflammation contributes to the atrial remodelling involving both structural and electrophysiological alterations that form the basis for the disease. A large-scale prospective study involving 24,734 women participants investigated the association of inflammatory markers such as CRP, fibrinogen, and intercellular adhesion molecule 1 (sICAM-1) with the incidence of AF. The results suggested that inflammation is a strong indicator for the incidence of AF with the median plasma levels of the biomarkers being independently correlated with the development of the disease in patients
[38][42]. That suggestion was further confirmed when scientists from Greece observed that the levels of high-sensitivity C-reactive protein (hs-CRP) are directly linked with the recurrence of AF after cardioversion and that the restoration of sinus rhythm (SR) resulted in a gradual decrease of hs-CRP
[39][43], while Rotter et al. reported that CRP levels in individuals with AF declined following effective ablation
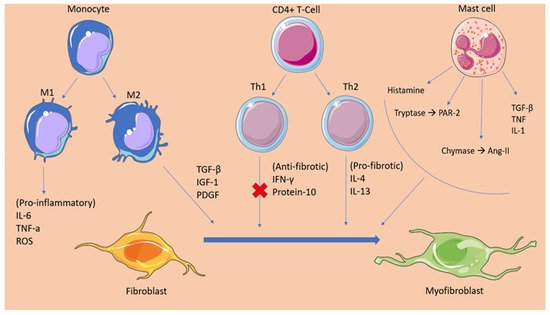
[40][41][44,45]. Additionally, in a recent study, Yao C. et al. demonstrated that, in patients with atrial fibrillation, the activity of NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome in atrial cardiomyocytes was considerably enhanced. The upregulation of the NLRP3 inflammasome promotes the release of damage-associated molecular patterns (DAMPs), which lead to the activation of cardiac fibroblasts, cells that, as described earlier, are the main effectors of cardiac fibrosis
[8][13].
Advances in the field of cardiology over the last years have led to the identification of many cellular and molecular mechanisms that suggest inflammation is responsible for the pathogenesis of AF. Under inflammatory stress, angiotensin II stimulates the production of proinflammatory cytokines (e.g., IL-6, IL-8, TNF-α) and the recruitment of immune cells. The role of AngII has also been established in the fibrosis and structural remodelling of the cardiac tissue through the activation of the MAPK-mediators of AngII/AT1R and the subsequent expression of the pro-fibrotic TGFβ1, which promotes fibroblast differentiation. Furthermore, increased pressure overload, as well as several gene polymorphisms in renin and angiotensin, mediate the formation of angiotensin II and the activation of angiotensin II receptors. Angiotensin II has been linked with the activation of NOX and the subsequent oxidation-related calcium-handling abnormalities, resulting in the electric remodelling of the atria. Additionally, NOX is a potent stimulator of the transcription factor nuclear factor-κB (NF-κB), which directly affects the sodium channel promoter regions, leading to a downregulation of the sodium channels and the promotion of AF mechanics
[42][43][46,47]. The RAAS system mechanism lying behind AF development reflects the theory that atrial fibrillation begets atrial fibrillation. This notion can be justified by recent evidence suggesting that AngII not only causes inflammation but also that inflammation can promote AngII production through hs-CRP and TNF-a. These molecules, which are pronounced in inflammatory states, seem to have an upregulatory effect on the AT1R, further promoting this vicious cycle
[44][48].
When associating inflammation with the occurrence of atrial fibrillation, it is important to mention the culprit of coronary artery disease in this phenomenon. Coronary heart disease has been associated with the development of atrial fibrillation through various mechanisms
[45][49]. Among them, inflammation constitutes the most important determinant of atrial fibrillation presentation, second only to atrial infarction and the subsequent tissue fibrosis. Following the event of myocardial ischemia, local as well as systemic inflammation arises, which causes the release of various inflammatory factors such as IL-6 and CRP, which have been independently associated with the development of atrial fibrillation
[46][50]. It has been proposed that IL-6 exerts its proarrhythmic effect by inducing atrial remodelling. Increased serum levels of IL-6 were associated by Psychari SN et al. with an increased left atrial size. The dilatation of the left atrium is believed to result from the stimulating effect of IL-6 on matrix-metalloproteinase-2 (MMP2), a protease that has been implicated in atrial remodeling
[47][51]. Moreover, it has been demonstrated that inflammation induced by myocardial infarction can promote atrial remodeling through the activation of Toll-like receptors (TLR), factors of the innate immune system. Particularly, TLR 2 and TLR 4 mRNA expression is significantly enhanced in patients following MI, while elevated TLR-2 levels have been associated with increased left atrial size
[48][49][52,53].
Of great importance when relating inflammation with AF, is the prothrombotic state present in the disease. A high CRP level has been related to the formation of thrombi in the left atrium
[50][54]. Research has established the mechanisms of thrombogenesis in inflammation. During an inflammatory state, innate immune cells activation and the release of inflammatory ligands are upregulated. IL-2, IL-6, IL-8, TNF-a, and MCP-1 production is enhanced by the activated immune cells resulting in the synthesis of tissue factor (TF), von Willebrand factor (vWF), and P-selectin
[51][55]. These molecules mediate platelet agglutination, as well as monocyte-endothelial cell attachment. This event combined with the endothelial damage induced in the atrium of a patient affected by AF severely increases the risk of thrombus formation
[52][53][54][56,57,58].