Over the past hundred years, atrial fibrillation (AF) is the arrhythmia that has been studied the most among all other heart rhythm disorders, leading to valuable conclusions [1]. The prevalence of AF ranges from 2% in the general population to 10–12% in those aged 80 and older [2]. It is the most common arrhythmia in humans, and incidence increases with advancing age [2]. According to the Global Burden of Disease, the estimated prevalence of AF is up to 33.5 million individuals, as it affects 2.5–3.5% of populations in several countries [3]. Atrial fibrosis has emerged as a significant pathophysiological component, with links to AF recurrences, resistance to medication, and complications [3].
Several mechanisms have been postulated to play a role in the development of AF, through both the electrical and structural remodeling of the atrial tissue. Among them, fibrosis has been studied thoroughly, confirming its significant role in this process.
Fibroses can been classified into two distinct types, reparative and interstitial fibrosis:
The two different types of fibrosis may coexist.
2.1. Cellular Mediators of Atrial Fibrosis
Several cellular subtypes have been investigated for their effect in the fibrotic process and the subsequent promotion of atrial fibrillation. Among them, fibroblasts have been established as the main cellular effectors of atrial fibrosis [
13]. Fibroblasts are small, spindle-shaped cells of mesenchymal origin, accounting for 10–15% of all cardiac tissue cells. [
14] They are metabolically active cells, regulating the synthesis and turnover of the ECM, thus preserving the architectural integrity of the cardiac tissue. Multiple communication pathways have been established between fibroblasts and cardiomyocytes, altering the latter’s electrophysiological properties. Under various pathological conditions and stress indicators, a phenotypic conversion of fibroblasts to alpha-smooth-muscle actin (αSMA) expressing myofibroblasts, takes place.
In detail, the activation and differentiation of local cardiac fibroblasts is dependent on multiple neurohumoral and mechanical profibrotic stress stimuli. Among the biochemical signals that have been identified to induce fibroblast differentiation, TGFβ has a prominent role in this process through both a canonical (SMAD-dependent) and non-canonical (SMAD-independent) pathway, which mediates the transcription of myofibroblast genes [
15,
16]. Additionally, angiotensin II (AngII) and endothelin 1 (ET-1), which bind to the G-protein-coupled receptors (GPCR) presented by cardiac fibroblasts, have been established as fibrotic mediators through the activation of a signaling cascade that promotes fibrotic gene transcription [
17]. The activation and differentiation of fibroblasts is further enhanced when mechanical forces are applied that generate a more tensile and rigid matrix. The mechanisms that have been proposed to be responsible for the tension-based induction of myofibroblasts rely either on the activation of stretch-sensitive transient receptor potential (TRP) channels, which further activate factors such as TGFβ, or the force-mediated activation of p38 from the contractile signals of the cytoskeleton [
18]. In conjunction with the aforementioned traditional fibroblast activation pathways, recent studies have brought to light significant mitochondrial, as well as cellular, metabolic components that promote the formation of myofibroblasts. Mitochondria act as key regulators in the fibroblast activation process by reducing their Ca
2+ uptake in response to the profibrotic signals, a process that further enhances the cytosolic Ca
2+ signaling pathway. Additionally, the profibrotic stressors induce the production of mitochondrial ROS, which activate factors such as p38 and ERK1/2, known for augmenting the transcription of fibrotic genes [
19]. Lastly, various cellular metabolic functions have been highlighted over the past few years among the main drivers of myofibroblast formation. In particular, an increase in the rate of glutaminolysis in fibroblasts is considered crucial for their activation, while alterations in glycolysis with the subsequent increase in lactate production have been proposed as essential mechanisms for the promotion of the myofibroblast differentiation program [
20]. Myofibroblast actions include recruiting inflammatory cells, promoting wound contraction, and secreting an excessive amount of ECM proteins such as collagen type I, III and IV; periostin; and fibronectin, leading to fibrosis [
21,
22].
In addition to fibroblasts, multiple inflammatory cells have been shown to be involved in the pro-fibrotic process. Studies have demonstrated the principal role that macrophages have in the regulation of fibrosis. Resident macrophages, originating from yolk sac-derived erythromyeloid progenitors (EMPs), populate the healthy myocardium, promoting its homeostasis. During the event of cardiac injury, multiple blood-borne monocytes infiltrate the myocardium and differentiate to macrophages [
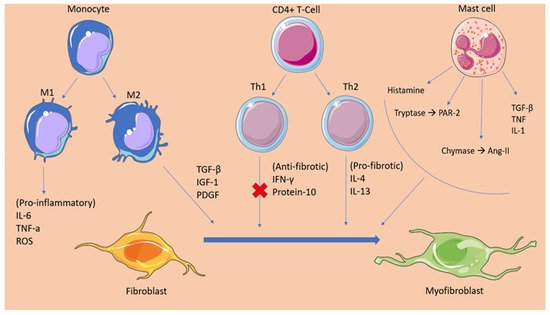
23]. Monocyte-derived macrophages express broad heterogeneity, enabling them to exert different functions, such as the production of multiple pro-fibrotic growth factors (IL-10, TGF-β, IGF-1, and PDGF), pro-inflammatory cytokines (IL-6, TNF-α, ROS), and proteases that contribute to matrix remodelling [
23].
Likewise, following myocardial injury, T-cells populate the cardiac tissue in response to cytokine signalling. T-cells are then differentiated into either CD4+ (Th1, Th2) or CD8+ cytotoxic T cells, which exert distinct functions. In the immediate post-insult period, Th1 and CD8+ cells are the main residents of the myocardium [
24]. These cells have been recognised for their anti-fibrotic functions, as they release mediators, such as IFN-γ and protein-10, which inhibit the action of the pro-fibrotic TGF-β. Additionally, INF-γ interferes with the activation of Th2 cells by impacting the production of IL4 and IL13 [
24]. Progressing into the chronic injury period, Th2 cells overtake Th1 cells as the principal CD4+ cell phenotype in the myocardial tissue. In contrast to the latter, Th2 cells exhibit significant pro-fibrotic activity. This is performed mainly by secreting IL4 and IL13, molecules that stimulate collagen secretion either by enabling TFG-β or by recruiting monocytes in the lesion site [
24].
Another component of the innate immunity, mast cells, have established their role as modulators for cardiac fibrosis. Studies have demonstrated that, under conditions of cardiac ischemia and pressure overload, mast cells multiplicate and degranulate pre-formed inflammatory and fibrotic (e.g.,TGF-β1, TNF, IL-1) mediators. Mast cells present in the cardiac tissue represent the connective tissue phenotype and contain both chymase and tryptase. Shiota et al. conducted a study that identified a 5.2-fold increase in chymase activity in hamsters with chronic pressure-overloaded hearts [
25]. Multiple studies have proven the pro-fibrotic effect of increased chymase activity in cardiac remodelling by promoting the formation of angiotensin-II [
26,
27,
28]. The increased levels of tryptase in fibrotic hearts have been shown to mediate fibroblast proliferation and differentiation to myofibroblasts. The mechanism responsible has been attributed to the stimulation of protease activated receptor-2 (PAR-2) in fibroblasts and the subsequent phosphorylation of extracellular signal-regulated protein kinases 1 and 2 (ERK ½), which promotes the differentiation of fibroblasts to myofibroblasts [
29]. Lastly, the role of histamine produced by mast cells has been thoroughly studied, establishing its significance in cardiac fibrosis. The detrimental role of histamine in cardiac fibrosis has been proven in an animal experiment, wherein a lack of histamine induced a response in H
2-receptor-deficient mice and reduced myocardial apoptosis and fibrosis [
30]. Nevertheless, multiple anti-inflammatory and anti-fibrotic mediators are also among the degranulation products of mast cells, raising controversy over the exact function of mast cells in the process of tissue remodelling [
31] (
Figure 1).
Figure 1. This graphical abstract summarizes the cellular mediators of atrial fibrosis. Following an insult, inflammatory mediators signal immune cells such as monocytes, CD4+ T-cells, and mast cells to infiltrate the atrial myocardium. These cells promote tissue fibrosis by secreting pro-fibrotic factors and regulatory molecules that enhance the activation and differentiation of fibroblasts to myofibroblasts. Additionally, the figure depicts the anti-fibrotic mediators that are secreted by Th1 cells in the early-insult stage and that are gradually overhauled by the products of pro-fibrotic Th2 cells. TGFβ, transforming growth factor beta; TNFα, tumor necrosis factor alpha; PDGF, platelet-derived growth factor; IL-1, interleukin 1; IL-4, interleukin 4; IL-6, interleukin 6; IL-10, interleukin 10; ROS, reactive oxygen species; IFNγ, interferon gamma; IGF-1, Insulin-like growth factor 1; Th1, t helper type 1; Th2, t helper type 2; PAR-2, protease activated receptor 2; Ang-II, angiotensin.
2.2. Fibrotic Mechanisms Inducing Atrial Fibrillation
Fibrosis has been established as a significant factor maintaining atrial fibrillation. There has been increased data associating the atrial remodelling induced by fibrosis with the promotion of AF. It has been proposed that the increased population of fibroblasts/myofibroblasts present in the fibrotic tissue and the increased deposition of ECM disrupt the myocardial bundles continuity, interfering with the gap-junction formation among cardiomyocytes. This event leads to conduction abnormalities, slowing conduction velocity and eventually forming unidirectional conduction blocks [
32]. Moreover, as mentioned previously, myofibroblasts form communication channels with cardiomyocytes, altering their electrophysiological properties, giving rise to focal firing and re-entrant circuits.
Over the last 10 years, several clinical studies have been conducted to confirm the aforementioned mechanisms. Researchers from the Cardiovascular Research institute in Maastricht performed epicardial mapping in 24 patients with long-standing persistent AF, undergoing cardiac surgery, in an attempt to uncover the spatiotemporal characteristics of the fibrillatory process underlying the disease. The study confirmed the intra-atrial conduction disturbances with the presence of block lines running in parallel to the muscular bundles [
33]. Additionally, a significant contribution in understanding the pathophysiology underlining the relationship between atrial fibrosis and arrhythmogenesis was made by Sebastien P.J Krul, et al., who studied the effect of interstitial fibrosis on conduction velocity [
34]. Researchers obtained 35 atrial appendages during AF surgery and recorded the activation time as well as the longitudinal (CVl) and transverse (CVt) conduction velocity (CV). The results demonstrated that the thick interstitial fibrotic strands were directly associated with an increase in the longitudinal CV in contrary to the transverse CV, which was not affected [
34]. However, a greater extent of transverse activation delay was observed because of the presence of activation block areas leading to a pattern of zig-zag conduction. This study points at the quality rather than the quantity of the fibrotic tissue as responsible for the formation of an arrhythmogenic substrate, with re-entry circuits enabling the perpetuation of atrial fibrillation [
34]. To further verify the driver mechanisms of AF, Hansen et al. performed a simultaneous mapping of the sub-endocardial and sub-epicardial activation patterns, and then integrated these data to an MRI-produced atrial model, in an attempt to visualize the AF drivers. The researchers confirmed the presence of longitudinal conduction blocks in agreement with the epicardial mapping study and, in addition, proved that fibrosis due to cardiac diseases disrupts the myocardial architecture, promoting a structural substrate for re-entrant AF drivers [
35].