Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christian Bailly | + 2284 word(s) | 2284 | 2021-12-20 04:21:42 | | | |
| 2 | Rita Xu | Meta information modification | 2284 | 2022-01-04 05:07:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bailly, C. Gal-9/TIM-3 Immune Checkpoint with α-Lactose. Encyclopedia. Available online: https://encyclopedia.pub/entry/17693 (accessed on 26 July 2026).
Bailly C. Gal-9/TIM-3 Immune Checkpoint with α-Lactose. Encyclopedia. Available at: https://encyclopedia.pub/entry/17693. Accessed July 26, 2026.
Bailly, Christian. "Gal-9/TIM-3 Immune Checkpoint with α-Lactose" Encyclopedia, https://encyclopedia.pub/entry/17693 (accessed July 26, 2026).
Bailly, C. (2022, January 01). Gal-9/TIM-3 Immune Checkpoint with α-Lactose. In Encyclopedia. https://encyclopedia.pub/entry/17693
Bailly, Christian. "Gal-9/TIM-3 Immune Checkpoint with α-Lactose." Encyclopedia. Web. 01 January, 2022.
Copy Citation
The disaccharide lactose is an excipient commonly used in pharmaceutical products. The two anomers, α- and β-lactose (α-L/β-L), differ by the orientation of the C-1 hydroxyl group on the glucose unit. In aqueous solution, a mutarotation process leads to an equilibrium of about 40% α-L and 60% β-L at room temperature.
lactose
galectin-9
PD-1/PD-L1
TIM-3
soluble PD-L1
1. Introduction
The modulation of cancer immunity represents the most potent therapeutic modality in oncology. Over the past ten years, the clinical approval of different immune checkpoint blockers has led to considerable improvement in the clinical outcome of cancer patients, notably those with solid tumors such as melanoma and lung cancer. However, for several other major cancers, death rates continue to increase, or the improvement progresses very slowly [1]. Research efforts must continue and novel approaches to improve prevention, early detection, and treatment are needed.
Novel immune checkpoint targets are proposed to offer new therapeutics and modalities to treat aggressive cancers such as pancreatic cancer, brain tumors, pleural mesothelioma, and triple-negative breast cancer for example [2][3][4][5][6][7]. Intense research efforts are also dedicated to improving the treatment of onco-hematological diseases. Targeting the immune system is considered a privileged axis to combat acute myeloid leukemia (AML) which is a highly aggressive disease, or to fight lymphoma in young patients, and multiple myeloma [8][9][10]. One of the emerging immune checkpoint molecules is the T-cell immunoglobulin mucin-3 (Tim-3)/Galectin-9 (Gal-9) checkpoint, viewed as a promising target for the treatment of breast cancer, bladder cancer and others [11][12][13][14]. Blockade of the TIM-3/Gal-9 checkpoint is considered a valid option in hematological malignancies, notably for the treatment of AML and myelodysplastic syndromes [15][16][17][18].
2. Lactose: α- and β-Anomers
Lactose is a disaccharide composed of a galactose unit linked to a glucose unit via a β-1,4 linkage (4-O-β-D-galactopyranosyl-D-glucopyranose, or Gal β(1→4) Glc). There are two anomers, α- and β-lactose, which differ in the configuration of the C-1 hydroxyl group of the glucose unit. α-Lactose (α-L) has an axial hydroxyl whereas the hydroxyl of β-lactose (β-L) is equatorial (Figure 1). The two anomers coexist in a solution, with an equilibrium of about 40% α-L and 60% β-L at room temperature. The α-anomer presents three crystalline forms: α-lactose monohydrate (α-LM), hygroscopic anhydrous α-lactose (α-LH) and stable anhydrous α-lactose (α-LS). The β-anomer (β-L) has one crystalline form [19][20]. The polymorphs of lactose exhibit distinct physical properties, in terms of solubility, density, stability, shelf life, dissolution rate, and bioavailability. α- and β-lactose differ considerably in solubility, β-L being much more water-soluble than α-L [21]. The interconversion between the different forms has been well investigated. During the mutarotation process, a short-lived hemiacetal intermediate is generated upon a ring-opening reaction of the glucose unit [22][23]. Similarly, the conversion of α-LM to the anhydrous forms is well-controlled [24]. The α/β lactose epimerization reaction occurs rapidly in aqueous solution (t1/2 = 28.3 min at 25 °C) and the product is stable [22]. An efficient method for the preparation of pure β-L from α-LM has been reported recently [25][26].
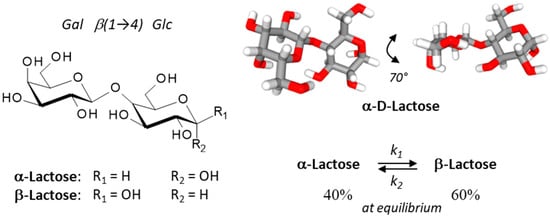
Figure 1. Structures of the two lactose anomers, α-lactose and β-lactose. Lactose contains a β-D-galactose unit linked to an α or a β-D-glucose unit through a β(1→4) bond. α-Lactose presents two crystalline forms, anhydrous α-lactose (C12H22O11) and α-lactose monohydrate (C12H22O11•H2O). The stability of lactose in aqueous solution has been determined [22]. An overall lactose epimerization rate constant (k) of 4.4 × 10−4 s−1 was measured at 25 °C (forward rate constant k1 of 2.5 × 10−4 s−1, and a reverse rate constant k2 of 1.5 ×10−4 s−1; K = 1.6 ± 0.1) and the half-life (t1/2) was 28.3 min at 25 °C. At equilibrium, the α/β anomeric ratio of lactose samples was about 39–41/59–61%, as determined at 25°, 45° and 60 °C.
Lactose is used as an additive in many foods and as a filler or diluent in pharmaceutical products. α-LM is an excipient commonly used for the manufacturing of dry granules and tablets. However, β-L anhydrous is also used in direct compression of tablet processes and as a filler in capsules [27]. As an excipient, lactose is present in nearly half of all solid medicines because it is readily available, cost-effective, chemically stable, and easily compatible with other excipients and active ingredients and has a bland taste. It provides a convenient matrix to stabilize different types of active pharmaceutical ingredients (API). α-Lactose monohydrate is also one of the most used carriers for dry powder inhaler formulations [28][29][30]. The lactose used as an excipient has generally a very high anomeric purity (about 96% α and 4% β) although the anomeric content of α-LM can vary significantly from one batch to another, and this variation can influence bioavailability from final formulations [31]. In solution, a rapid lactose epimerization process can occur [22]. Dehydration leads to solid-state epimerization in α-lactose powders [32]. The form of lactose can be extremely important as it can serve to modulate the property of the active ingredient. For example, the use of highly porous lactose as a carrier can significantly improve the solubility of the flavonoid quercetin and its release, compared to a conventional α-lactose carrier [33]. The lactose excipient can influence the dissolution properties of the API present in the drug tablet, notably to improve the dissolution behavior of an insoluble API. Different types of lactose-based excipients and co-excipients are available [34][35].
Lactose is considered a pharmacologically inactive substance, which is the definition of an excipient. However, lactose is not devoid of biological activity. People intolerant to lactose may experience severe clinical symptoms (such as diarrhea and bloating); the disease severity varies considerably among individuals. The intolerance is generally due to the absence or deficiency of the lactase enzyme (lactase-phlorizin hydrolase) found in the small intestinal brush border, which hydrolyses lactose into glucose and galactose. Lactose intolerance is a relatively common gastrointestinal condition, at least in children, caused by the inability to digest and absorb dietary lactose [36][37]. Lactose can serve as an inducer of protein synthesis in cell cultures [38] and can induce senescence in human lung fibroblasts [39]. Moreover, in recent years the immunological functions of lactose have been delineated, notably its role as a galectin ligand and more specifically as a key regulator of the TIM-3 receptor. The present review highlights the importance of lactose as a checkpoint regulator.
3. The TIM-3 Immune Checkpoint and Its Targeting with Antibodies
Monoclonal antibodies (mAbs) targeting critical immune checkpoints, such as PD-1/PD-L1 and CTLA-4, have significantly ameliorated the treatment of advanced solid tumors, such as lung (NSCLC), skin and liver cancers. Immune checkpoint inhibitors targeting and blocking the interaction of certain cell surface proteins, such as PD-1 expressed on diverse immune cells or PD-L1 expressed on cancer cells, act as brakes on immune responses [40][41]. A handful of mAbs directed against PD-1 or PD-L1 have been approved for cancer treatment, including pembrolizumab, nivolumab, atezolizumab, camrelizumab and others. Often combined with chemo- or radiotherapy, these mAbs have provided significant survival benefits in cancer patients [42]. However, not all tumors are sufficiently sensitive to anti-PD-1/PD-L1 therapies and durable responses are not always achieved. Therefore, novel immunotherapeutic approaches are needed to combat advanced or relapsed cancers. As for PD-1, other immune checkpoints commonly associated with T cells such as LAG-3 and TIM-3 have been considered [43][44].
T cell immunoglobulin and mucin domain molecule 3 (TIM-3) is a protein now well exploited as a therapeutic target for cancer immunotherapy [45][46][47][48]. This co-inhibitory receptor is expressed on CD4+ and CD8+ T cells which produce interferon-γ (IFN-γ) and on specific innate immune cells (Figure 2). In addition, the expression of membrane TIM-3 has been evidenced at the surface of cancer cells, such as renal, lung, gastric, prostate, and other types of cancer cells. TIM-3 plays major roles in the regulation of tumorigenesis, inflammation, and antitumor immunity [49]. Importantly, TIM-3 is upregulated on CD8+ T cells and Tregs, in tumors treated with radiotherapy and anti-PD-L1 antibodies. The treatment with an anti-TIM-3 mAb concurrently with anti-PD-L1 and radiotherapy induced a significant tumor growth delay, an enhancement of T-cell cytotoxicity, a decrease of Tregs, and improved survival in models of head and neck squamous cell carcinoma [50]. The data suggested that the blockade of Tim-3 can antagonize the resistance of PD-1/PD-L1 blocking therapy [51]. Major anticancer effects have been reported in cases of Tim-3 blockade, in different types of experimental tumors, incusing breast cancer [12][52], hepatocellular carcinoma [53], and other solid tumors [54]. The blockade of TIM-3 is also a promising approach for the treatment of hematological malignancies, including myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) [17][55][56][57]. In AML, studies have shown an overexpression of TIM-3 on leukemia stem cells (LSC) but not on healthy stem cells [58][59][60]. The blockade of the receptor induces a direct inhibition of AML cell proliferation and restores T cell function [16]. Thus, targeting TIM-3 offers a unique opportunity to target the LSC population resistant to chemotherapy and largely implicated in tumor relapse [61].
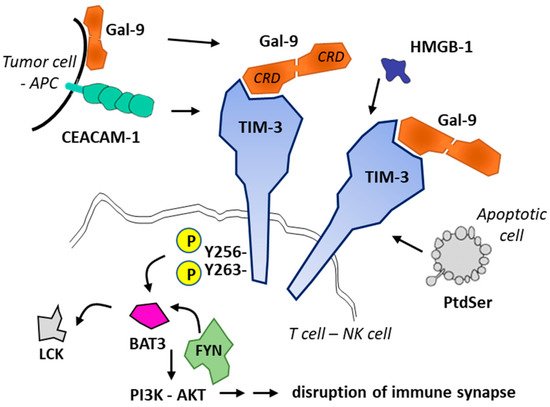
Figure 2. Ligands and signaling of TIM-3. Four TIM-3 ligands have been identified: CEACAM1, phosphatidylserine (PtdSer released from apoptotic cells), HMGB-1 and Gal-9. Gal-9 can be secreted by antigen-presenting cells (APCs) or tumor cells or present at the cell surface. Gal-9 (with two carbohydrate recognition domains (CRD) separated by a short linker) promotes oligomerization of TIM3 and triggers signaling via phosphorylation of residues Tyr256 and Tyr263 in the intracellular domain of TIM3. The phosphorylation releases the adaptor protein BAT3 (HLA-B-associated transcript 3) and allows recruitment of tyrosine kinase FYN, whereas in its ligand-unbound form, TIM-3 interacts with BAT3 and recruits the kinase LCK to maintain T cell activation. Fyn and Bat3 are two adaptor molecules involved in inhibition and activation of Tim-3 downstream signaling, respectively. Gal-9-mediated recruitment of FYN leads to the disruption of immune synapse formation and to cell apoptosis [45].
There are several anti-TIM-3 monoclonal antibodies in (pre)clinical development [62], such as (i) sabatolimab which is currently evaluated for the treatment of advanced solid tumors [63] and myelodysplastic syndromes [64], (ii) the fully human anti-TIM-3 antibody M6903 in preclinical development [65], (iii) LY3321367 currently tested in advanced solid tumors [66], and a few other antibodies [49][67] (Table 1). Over the past two years, TIM-3 has emerged as an important immune-checkpoint molecule and recent data demonstrated that the receptor plays multiple roles, well beyond CD4+/CD8+ T cells, notably as a key regulator of dendritic cell functions via the regulation of inflammasome activation [68]. The discovery of the many functions of the TIM-3 receptor has encouraged the design and development of different therapeutic entities, monoclonal antibodies but also anti-TIM-3 chimeric antigen receptor (CAR)-directed T lymphocyte therapy [44] and anti-TIM-3 oligonucleotide aptamers [69][70] (Figure 3).
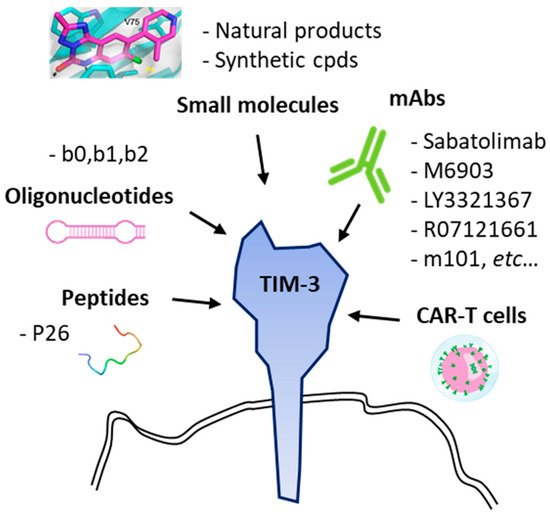
Table 1. Human anti-TIM-3 antibodies in development.
| Anti-TIM-3 Antibodies | Current Development | References |
|---|---|---|
| LY3321367 (Eli Lilly; Indianapolis, IN, USA) |
Phase 1 trial in patients with relapsed/refractory solid tumor, alone or in combination with an anti-PD-L1 mAb. NCT03099109 * | [66][72] |
| Sabatolimab (MBG453, Novartis, Basel, Switzerland) |
Fast track designation by US FDA for the treatment of patients with myelodysplastic syndromes (MDS) | [73] |
| Phase 1 trial in patients with advanced solid tumor, alone or in combination with anti-PD-1 spartalizumab. | [63] | |
| Phase 1 trial in patients with Acute Myeloid Leukemia (AML). NCT04812548, NCT04623216, NCT04878432, NCT04150029 * | [74] | |
| BMS-986258 (ONO 7807, BMS, Lawrenceville, NJ, USA) |
Fully human anti-TIM-3 mAb, tested in combination with anti-PD-1 nivolumab in patients with advanced solid tumors (Phase 1). NCT03446040 * | |
| Cobolimab (TSR-022, GSK, Brentford, UK) |
Anti-TIM-3 mAb in combination with anti-PD-1 in patients with liver cancer or with a melanoma (Phase 1). NCT04655976, NCT03680508, NCT04139902, NCT02817633 * | |
| Sym023 (Symphogen; Ballerup, Denmark) |
Fully human anti-TIM-3 mAb, in patients with advanced solid tumor malignancies or lymphomas (Phase 1). NCT03489343 * | |
| INCAGN02390 (Incyte, Agenus; Wilmington, DE, USA) |
Phase 1 study to determine the safety, tolerability, and preliminary efficacy in participants with advanced malignancies. Fully human Fc-engineered IgG1k. NCT03652077, NCT04370704. | [75] |
| RO7121661 (Roche; Basel, Switzerland) |
Anti-PD-1/TIM-3 bispecific mAbs, tested in patients with advanced and/or metastatic solid tumors (Phase 1). NCT03708328 * | |
| BGB-A425 (BeiGene Ltd., Beijing, China) |
Humanized anti-TIM-3 mAb, in combination with anti-PD-1 mAb tislelizumab, in patients with advanced solid tumors (phase I/II trial). NCT03744468 * | [76] |
| M6903 | Fully human anti-TIM-3 mAb, without effector function, which blocks binding of TIM-3 to the 3 ligands phosphatidylserine, CEACAM1, and Gal-9. Experimental laboratories studies. | [65] |
| F38.2E2 | Anti-human TIM-3 antibody capable of blocking binding of TIM-3 to phosphatidylserine and CEACAM1. Experimental tool. | [77] |
* ClinicalTrials.gov Identifier.
Anti-TIM-3 biotherapeutic agents have demonstrated potent efficacy in preclinical models and the first clinical data obtained with sabatolimab look promising for the treatment of MDS [78]. A phase 2 trial is ongoing in MDS with sabatolimab combined with azacitidine and venetoclax (NCT04812548). Combined with azacytidine, the anti-TIM-3 antibody might turn to be also useful for the treatment refractory acute myeloid leukemia (AML). A major phase 1–2 trial is currently ongoing (NCT04623216). However, the challenge remains high at present, as no definite proof of long-term clinical efficacy has been reported in AML or MDS. In CD8+ T cells collected from patients with chronic lymphocytic leukemia (CLL), the blockade of an anti-TIM-3 antibody has failed to restore the function of these exhausted T cells [79]. Moreover, a study has shown that the treatment with anti-TIM-3 mAb can inhibit the phagocytic ability of alveolar macrophages in the lungs of mice and thus might cause pneumonitis [80]. The biological and clinical consequences of TIM-3 blockade are not completely understood at present.
Therapeutic antibodies and engineered cells targeting TIM-3 have been designed and are being tested. However, to our knowledge, no small molecules specifically targeting TIM-3 have been clinically developed as yet, despite the potential benefits of the targeting approach in oncology and in other therapeutic fields [14]. The TIM-3-associated immune dysregulation mechanisms of CD4+ or CD8+ T cells have been associated with different inflammatory diseases such as vitiligo [81][82], rheumatoid arthritis [83][84], liver diseases [85] and in viral infections such as human papilloma virus (HPV) positive cervical cancer [86]. There is a need for small molecules targeting TIM-3, to reduce the cost of treatment compared to mAbs, to facilitate drug combinations and to allow oral treatments. Thus far, the development of small molecules lags far behind the development of small molecules, as discussed below.
References
- Islami, F.; Ward, E.M.; Sung, H.; Cronin, K.A.; Tangka, F.K.L.; Sherman, R.L.; Zhao, J.; Anderson, R.N.; Henley, S.J.; Yabroff, K.R.; et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. J. Natl. Cancer Inst. 2021, 113, 1648–1669.
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138.
- Sukumar, J.; Gast, K.; Quiroga, D.; Lustberg, M.; Williams, N. Triple-negative breast cancer: Promising prognostic biomarkers currently in development. Expert Rev. Anticancer Ther. 2021, 21, 135–148.
- Desbaillets, N.; Hottinger, A.F. Immunotherapy in Glioblastoma: A Clinical Perspective. Cancers 2021, 13, 3721.
- Corti, C.; Nicolò, E.; Curigliano, G. Novel immune targets for the treatment of triple-negative breast cancer. Expert Opin. Ther. Targets. 2021, 25, 815–834.
- Tang, D.; Lotze, M.T. Tumor immunity times out: TIM-3 and HMGB1. Nat. Immunol. 2012, 13, 808–810.
- Lee, J.B.; Ha, S.J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 681320.
- Iżykowska, K.; Rassek, K.; Korsak, D.; Przybylski, G.K. Novel targeted therapies of T cell lymphomas. J. Hematol. Oncol. 2020, 13, 176.
- Fink, A.; Hung, E.; Singh, I.; Ben-Neriah, Y. Immunity in acute myeloid leukemia: Where the immune response and targeted therapy meet. Eur. J. Immunol. Online ahead of print. 2021.
- Krejcik, J.; Barnkob, M.B.; Nyvold, C.G.; Larsen, T.S.; Barington, T.; Abildgaard, N. Harnessing the Immune System to Fight Multiple Myeloma. Cancers 2021, 13, 4546.
- Mohsenzadegan, M.; Bavandpour, P.; Nowroozi, M.R.; Amini, E.; Kourosh-Arami, M.; Momeni, S.A.; Bokaie, S.; Sharifi, L. The Potential of T Cell Immunoglobulin and Mucin-Domain containing-3 (Tim-3) in Designing Novel Immunotherapy for Bladder Cancer. Endocr. Metab. Immune Disord. Drug Targets. Online ahead of print. 2021.
- Cong, Y.; Liu, J.; Chen, G.; Qiao, G. The Emerging Role of T-Cell Immunoglobulin Mucin-3 in Breast Cancer: A Promising Target For Immunotherapy. Front. Oncol. 2021, 11, 723238.
- Zang, K.; Hui, L.; Wang, M.; Huang, Y.; Zhu, X.; Yao, B. TIM-3 as a Prognostic Marker and a Potential Immunotherapy Target in Human Malignant Tumors: A Meta-Analysis and Bioinformatics Validation. Front. Oncol. 2021, 11, 579351.
- Zeidan, A.M.; Komrokji, R.S.; Brunner, A.M. TIM-3 pathway dysregulation and targeting in cancer. Expert Rev. Anticancer Ther. 2021, 21, 523–534.
- Asayama, T.; Taura, H.; Ishibashi, M.; Kuribayashi-Hamada, Y.; Onodera-Kondo, A.; Okuyama, N.; Yamada, A.; Shimizu, M.; Moriya, K.; Takahashi, H.; et al. Functional expression of Tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget 2017, 8, 88904–88917.
- Rezaei, M.; Tan, J.; Zeng, C.; Li, Y.; Ganjalikhani-Hakemi, M. TIM-3 in Leukemia; Immune Response and Beyond. Front. Oncol. 2021, 11, 753677.
- Wang, Z.; Chen, J.; Wang, M.; Zhang, L.; Yu, L. One Stone, Two Birds: The Roles of Tim-3 in Acute Myeloid Leukemia. Front Immunol. 2021, 12, 618710.
- Daver, N. Immune checkpoint inhibitors in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2021, 34, 101247.
- Platteau, C.; Lefebvre, J.; Affouard, F.; Derollez, P. Ab initio structure determination of the hygroscopic anhydrous form of alpha-lactose by powder X-ray diffraction. Acta Crystallogr. B 2004, 60, 453–460.
- Platteau, C.; Lefebvre, J.; Affouard, F.; Willart, J.F.; Derollez, P.; Mallet, F. Structure determination of the stable anhydrous phase of alpha-lactose from X-ray powder diffraction. Acta Crystallogr. B 2005, 61, 185–191.
- Xiang, J.; Wang, B.; Fu, L.; Chen, C.; Liu, W.; Tan, S. Tailoring α/β Ratio of Pollen-Like Anhydrous Lactose as Ingredient Carriers for Controlled Dissolution Rate. Crystals 2021, 11, 1049.
- Jawad, R.; Drake, A.F.; Elleman, C.; Martin, G.P.; Warren, F.J.; Perston, B.B.; Ellis, P.R.; Hassoun, M.A.; Royall, P.G. Stability of sugar solutions: A novel study of the epimerization kinetics of lactose in water. Mol. Pharm. 2014, 11, 2224–2238.
- Chen, Z.; Wu, T.; Yang, X.; Yue, F.; Fu, F. An exploration of the solvent- and acid-catalyzed mutarotation mechanisms of lactose in aqueous solution. New J. Chem. 2020, 44, 16421–16430.
- Batra, A.; Desai, D.; Serajuddin, A.T.M. Conversion of alpha-lactose monohydrate to anhydrous form with superior tabletability by twin-screw extrusion at elevated temperature. Int. J. Pharm. 2020, 588, 119790.
- López-Pablos, A.L.; Leyva-Porras, C.C.; Silva-Cázares, M.B.; Longoria-Rodríguez, F.E.; Pérez-García, S.A.; Vértiz-Hernández, A.A.; Saavedra-Leos, M.Z. Preparation and Characterization of High Purity Anhydrous β-Lactose from α-Lactose Monohydrate at Mild Temperature. Int. J. Polym. Sci. 2018, 2018, 5069063.
- Lara-Mota, E.E.; Nicolás-Vázquez, M.I.; López-Martínez, L.A.; Espinosa-Solis, V.; Cruz-Alcantar, P.; Toxqui-Teran, A.; Saavedra-Leos, M.Z. Phenomenological study of the synthesis of pure anhydrous beta-lactose in alcoholic solution. Food Chem. 2021, 340, 128054.
- EMA/CHMP/186428/2016. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Information for the Package Leaflet Regarding Lactose used as an Excipient in Medicinal Products for Human Use. Public Consultation: 19 November 2018–31 May 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-information-package-leaflet-regarding-lactose-used-excipient-medicinal-products-human-use_en.pdf (accessed on 12 October 2021).
- Della Bella, A.; Müller, M.; Soldati, L.; Elviri, L.; Bettini, R. Quantitative determination of micronization-induced changes in the solid state of lactose. Int. J. Pharm. 2016, 505, 383–393.
- Mehta, P. Imagine the Superiority of Dry Powder Inhalers from Carrier Engineering. J. Drug Deliv. 2018, 2018, 5635010.
- Ke, W.R.; Chang, R.Y.K.; Kwok, P.C.L.; Chen, D.; Chan, H.K. Spray drying lactose from organic solvent suspensions for aerosol delivery to the lungs. Int. J. Pharm. 2020, 591, 119984.
- Altamimi, M.J.; Wolff, K.; Nokhodchi, A.; Martin, G.P.; Royall, P.G. Variability in the α and β anomer content of commercially available lactose. Int. J. Pharm. 2019, 555, 237–249.
- Alzoubi, T.; Martin, G.P.; Barlow, D.J.; Royall, P.G. Stability of alpha-lactose monohydrate: The discovery of dehydration triggered solid-state epimerization. Int. J. Pharm. 2021, 604, 120715.
- Liu, W.; Wang, T.T.; Tang, X.L.; Jiang, F.Y.; Yan, X.; Deng, J. Porous Lactose as a Novel Ingredient Carrier for the Improvement of Quercetin Solubility In Vitro. Bioinorg. Chem. Appl. 2021, 2021, 2586990.
- Dominik, M.; Vraníková, B.; Svačinová, P.; Elbl, J.; Pavloková, S.; Prudilová, B.B.; Šklubalová, Z.; Franc, A. Comparison of Flow and Compression Properties of Four Lactose-Based Co-Processed Excipients: Cellactose® 80, CombiLac®, MicroceLac® 100, and StarLac®. Pharmaceutics 2021, 13, 1486.
- Zellnitz, S.; Lamešić, D.; Stranzinger, S.; Pinto, J.T.; Planinšek, O.; Paudel, A. Spherical agglomerates of lactose as potential carriers for inhalation. Eur. J. Pharm. Biopharm. 2021, 159, 11–20.
- Di Costanzo, M.; Biasucci, G.; Maddalena, Y.; Di Scala, C.; De Caro, C.; Calignano, A.; Canani, R.B. Lactose Intolerance in Pediatric Patients and Common Misunderstandings About Cow’s Milk Allergy. Pediatr. Ann. 2021, 50, 178–185.
- Catanzaro, R.; Sciuto, M.; Marotta, F. Lactose intolerance: An update on its pathogenesis, diagnosis, and treatment. Nutr. Res. 2021, 89, 23–34.
- Zhang, L.; Chen, X.; Deng, Y.; Jiang, C.; Deng, N. Fermentation, purification and immunogenicity of a recombinant tumor multi-epitope vaccine, VBP3. Protein Expr. Purif. 2020, 174, 105658.
- Xing, S.; Zhang, L.; Lin, H.; Mao, Z.; Bao, K.; Hao, P.; Pei, Z.; Li, J.; Hu, Z. Lactose induced redox-dependent senescence and activated Nrf2 pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 2034–2045.
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front. Pharmacol. 2021, 12, 731798.
- Hosseinzadeh, R.; Feizisani, F.; Shomali, N.; Abdelbasset, W.K.; Hemmatzadeh, M.; Gholizadeh, J.; Jadidi-Niaragh, F.; Bokov, D.O.; Janebifam, M.; Mohammadi, H. PD-1/PD-L1 Blockade: Prospectives for immunotherapy in Cancer and Autoimmunity. IUBMB Life. 2021, 73, 1293–1306.
- Bailly, C.; Thuru, X.; Quesnel, B. Combined cytotoxic chemotherapy and immunotherapy of cancer: Modern times. NAR Cancer 2020, 2, zcaa002.
- Dempke, W.C.M.; Fenchel, K.; Uciechowski, P.; Dale, S.P. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur. J. Cancer 2017, 74, 55–72.
- Lee, W.S.; Ye, Z.; Cheung, A.M.S.; Goh, Y.P.S.; Oh, H.L.J.; Rajarethinam, R.; Yeo, S.P.; Soh, M.K.; Chan, E.H.L.; Tan, L.K.; et al. Effective Killing of Acute Myeloid Leukemia by TIM-3 Targeted Chimeric Antigen Receptor T Cells. Mol. Cancer Ther. 2021, 20, 1702–1712.
- Joller, N.; Kuchroo, V.K. Tim-3, Lag-3, and TIGIT. Emerg. Concepts Target. Immune Checkp. Cancer Autoimmun. 2017, 410, 127–156.
- Saleh, R.; Toor, S.M.; Elkord, E. Targeting TIM-3 in solid tumors: Innovations in the preclinical and translational realm and therapeutic potential. Expert Opin. Ther. Targets. 2020, 24, 1251–1262.
- Kandel, S.; Adhikary, P.; Li, G.; Cheng, K. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett. 2021, 510, 67–78.
- Zhao, L.; Cheng, S.; Fan, L.; Zhang, B.; Xu, S. TIM-3: An update on immunotherapy. Int. Immunopharmacol. 2021, 99, 107933.
- Cao, Y.; Li, Q.; Liu, H.; He, X.; Huang, F.; Wang, Y. Role of Tim-3 in regulating tumorigenesis, inflammation, and antitumor immunity therapy. Cancer Biomark. 2021, 32, 237–248.
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380.
- Tian, T.; Li, Z. Targeting Tim-3 in Cancer With Resistance to PD-1/PD-L1 Blockade. Front. Oncol. 2021, 11, 731175.
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selnø, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Gonçalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594.
- Yu, L.; Liu, X.; Wang, X.; Yan, F.; Wang, P.; Jiang, Y.; Du, J.; Yang, Z. TIGIT(+) TIM-3(+) NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virus-related hepatocellular carcinoma. Oncoimmunology 2021, 10, 1942673.
- Solinas, C.; De Silva, P.; Bron, D.; Willard-Gallo, K.; Sangiolo, D. Significance of TIM3 expression in cancer: From biology to the clinic. Semin. Oncol. 2019, 46, 372–379.
- Gonçalves Silva, I.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Siligardi, G.; Ceccone, G.; et al. The Tim-3-galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine 2017, 22, 44–57.
- Tao, J.; Han, D.; Gao, S.; Zhang, W.; Yu, H.; Liu, P.; Fu, R.; Li, L.; Shao, Z. CD8+ T cells exhaustion induced by myeloid-derived suppressor cells in myelodysplastic syndromes patients might be through TIM3/Gal-9 pathway. J. Cell. Mol. Med. 2020, 24, 1046–1058.
- Dama, P.; Tang, M.; Fulton, N.; Kline, J.; Liu, H. Gal9/Tim-3 expression level is higher in AML patients who fail chemotherapy. J. Immunother. Cancer 2019, 7, 175.
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010, 7, 708–717.
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell 2015, 17, 341–352.
- Kikushige, Y.; Miyamoto, T. Identification of TIM-3 as a Leukemic Stem Cell Surface Molecule in Primary Acute Myeloid Leukemia. Oncology 2015, 89, 28–32.
- Marchand, T.; Pinho, S. Leukemic Stem Cells: From Leukemic Niche Biology to Treatment Opportunities. Front. Immunol. 2021, 12, 775128.
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911.
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629.
- Bewersdorf, J.P.; Zeidan, A.M. Management of patients with higher-risk myelodysplastic syndromes after failure of hypomethylating agents: What is on the horizon? Best Pract. Res. Clin. Haematol. 2021, 34, 101245.
- Zhang, D.; Jiang, F.; Zaynagetdinov, R.; Huang, H.; Sood, V.D.; Wang, H.; Zhao, X.; Jenkins, M.H.; Ji, Q.; Wang, Y.; et al. Identification and characterization of M6903, an antagonistic anti-TIM-3 monoclonal antibody. Oncoimmunology 2020, 9, 1744921.
- Harding, J.J.; Moreno, V.; Bang, Y.J.; Hong, M.H.; Patnaik, A.; Trigo, J.; Szpurka, A.M.; Yamamoto, N.; Doi, T.; Fu, S.; et al. Blocking TIM-3 in Treatment-refractory Advanced Solid Tumors: A Phase Ia/b Study of LY3321367 with or without an Anti-PD-L1 Antibody. Clin. Cancer Res. 2021, 27, 2168–2178.
- Jin, Y.J.; Yu, D.; Tian, X.L.; Li, H.X.; Zhou, X.C.; Kong, Y.; Zhang, W.; Zhang, L.; Lei, C.; Yang, Z.L.; et al. A novel and effective approach to generate germline-like monoclonal antibodies by integration of phage and mammalian cell display platforms. Acta Pharmacol. Sin. 2021, 1–9.
- Dixon, K.O.; Tabaka, M.; Schramm, M.A.; Xiao, S.; Tang, R.; Dionne, D.; Anderson, A.C.; Rozenblatt-Rosen, O.; Regev, A.; Kuchroo, V.K. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 2021, 595, 101–106.
- Gefen, T.; Castro, I.; Muharemagic, D.; Puplampu-Dove, Y.; Patel, S.; Gilboa, E. A TIM-3 Oligonucleotide Aptamer Enhances T Cell Functions and Potentiates Tumor Immunity in Mice. Mol. Ther. 2017, 25, 2280–2288.
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185.
- Zhong, T.; Zhao, C.; Wang, S.; Tao, D.; Ma, S.; Shou, C. The biologically functional identification of a novel TIM3-binding peptide P26 in vitro and in vivo. Cancer Chemother. Pharmacol. 2020, 86, 783–792.
- Hollebecque, A.; Chung, H.C.; de Miguel, M.J.; Italiano, A.; Machiels, J.P.; Lin, C.C.; Dhani, N.C.; Peeters, M.; Moreno, V.; Su, W.C.; et al. Safety and Antitumor Activity of α-PD-L1 Antibody as Monotherapy or in Combination with α-TIM-3 Antibody in Patients with Microsatellite Instability-High/Mismatch Repair-Deficient Tumors. Clin. Cancer Res. 2021, 27, 6393–6404.
- Zeidan, A.M.; Al-Kali, A.; Borate, U.; Cluzeau, T.; DeZern, A.E.; Giagounidis, A.; Kobata, K.; Lyons, R.; Platzbecker, U.; Sallman, D.A.; et al. Sabatolimab (MBG453) Combination Treatment Regimens for Patients (Pts) with Higher-Risk Myelodysplastic Syndromes (HR-MDS): The MDS Studies in the Stimulus Immuno-Myeloid Clinical Trial Program. Blood 2021, 138, 4669.
- Bruner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated Results from a Phase 1b Study. Blood 2020, 136, 1–2.
- Waight, J.; Iyer, P.; Breous-Nystrom, E.; Riordan, C.; Findeis, M.; Underwood, D.; Connolly, J.; Sanicola-Nadel, M.; Nastri, H.; Scherle, P.; et al. Abstract 3825: INCAGN02390, a novel antagonist antibody that targets the co-inhibitory receptor TIM-3. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018; 2018; 78, p. 3825.
- Desai, J.; Meniawy, T.; Beagle, B.; Li, Z.Z.; Mu, S.; Wu, J.; Denlinger, C.S.; Messersmith, W.A. Bgb-A425, an investigational anti-TIM-3 monoclonal antibody, in combination with tislelizumab, an anti-PD-1 monoclonal antibody, in patients with advanced solid tumors: A phase I/II trial in progress. J. Clin. Oncol. 2020, 38, TPS3146.
- Sabatos-Peyton, C.A.; Nevin, J.; Brock, A.; Venable, J.D.; Tan, D.J.; Kassam, N.; Xu, F.; Taraszka, J.; Wesemann, L.; Pertel, T.; et al. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology 2017, 7, e1385690.
- Santini, V. Advances in myelodysplastic syndrome. Curr. Opin. Oncol. 2021, 33, 681–686.
- Rezazadeh, H.; Astaneh, M.; Tehrani, M.; Hossein-Nataj, H.; Zaboli, E.; Shekarriz, R.; Asgarian-Omran, H. Blockade of PD-1 and TIM-3 immune checkpoints fails to restore the function of exhausted CD8(+) T cells in early clinical stages of chronic lymphocytic leukemia. Immunol. Res. 2020, 68, 269–279.
- Isshiki, T.; Akiba, H.; Nakayama, M.; Harada, N.; Okumura, K.; Homma, S.; Miyake, S. Cutting Edge: Anti-TIM-3 Treatment Exacerbates Pulmonary Inflammation and Fibrosis in Mice. J. Immunol. 2017, 199, 3733–3737.
- Tembhre, M.K.; Parihar, A.S.; Sharma, A.; Gupta, S.; Chattopadhyay, P.; Sharma, V.K. Participation of T cell immunoglobulin and mucin domain-3 (TIM-3) and its ligand (galectin-9) in the pathogenesis of active generalized vitiligo. Immunol. Res. 2015, 62, 23–34.
- Rahimi, A.; Hossein-Nataj, H.; Hajheydari, Z.; Aryanian, Z.; Shayannia, A.; Ajami, A.; Asgarian-Omran, H. Expression analysis of PD-1 and Tim-3 immune checkpoint receptors in patients with vitiligo; positive association with disease activity. Exp. Dermatol. 2019, 28, 674–681.
- Koohini, Z.; Hossein-Nataj, H.; Mobini, M.; Hosseinian-Amiri, A.; Rafiei, A.; Asgarian-Omran, H. Analysis of PD-1 and Tim-3 expression on CD4+ T cells of patients with rheumatoid arthritis; negative association with DAS28. Clin. Rheumatol. 2018, 37, 2063–2071.
- Matsumoto, H.; Fujita, Y.; Asano, T.; Matsuoka, N.; Temmoku, J.; Sato, S.; Yashiro-Furuya, M.; Watanabe, H.; Migita, K. T cell immunoglobulin and mucin domain-3 is associated with disease activity and progressive joint damage in rheumatoid arthritis patients. Medicine 2020, 99, e22892.
- Zhao, L.; Yu, G.; Han, Q.; Cui, C.; Zhang, B. TIM-3: An emerging target in the liver diseases. Scand. J. Immunol. 2020, 91, e12825.
- Chen, Z.; Dong, D.; Zhu, Y.; Pang, N.; Ding, J. The role of Tim-3/Galectin-9 pathway in T-cell function and prognosis of patients with human papilloma virus-associated cervical carcinoma. FASEB J. 2021, 35, e21401.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
04 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No