 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Uday Kishore | + 3143 word(s) | 3143 | 2021-12-27 09:07:23 | | | |
| 2 | Lindsay Dong | Meta information modification | 3143 | 2021-12-30 10:44:41 | | |
Video Upload Options
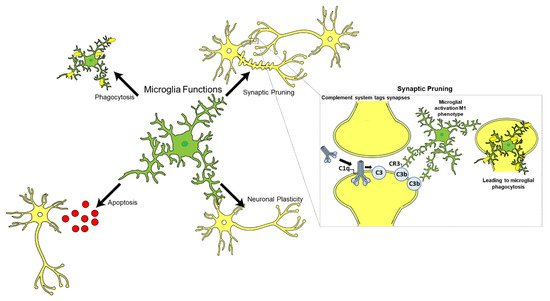
Alzheimer’s disease is a type of dementia characterized by problems with short-term memory, cognition, and difficulties with activities of daily living. It is a progressive, neurodegenerative disorder. The complement system is an ancient part of the innate immune system and comprises of more than thirty serum and membrane-bound proteins. This system has three different activating pathways and culminates into the formation of a membrane attack complex that ultimately causes target cell lysis (usually pathogens) The complement system is involved in several important functions in the central nervous system (CNS) that include neurogenesis, synaptic pruning, apoptosis, and neuronal plasticity.
1. Introduction
2. Role of the Complement System in CNS
The Complement System
| Functions of Complement System | Role | Mechanism | Reference |
|---|---|---|---|
| Neuroprotection | Neurogenesis | Increased complement receptor activation in the development of cerebellar neurons in animal models. | [17] |
| Disrupting C3aR signalling in mice models impairs neurogenesis. | [18] | ||
| CR2 is a negative regulator of neurogenesis. | [17][18] | ||
| Synaptic pruning | C1q−/− mice exhibit increased synaptic connections resulting in epilepsy, indicating an essential role in synaptic pruning. | [19][20] | |
| Synaptic plasticity | C1q−/− mice show weak dendrites and spines. | [19] | |
| Neuroinflammation | Binding with Aβ | Activation of classical pathway. | [9][10][21] |
| Binding with Tau protein | Activation of complement system via classical pathway. | [12][22] | |
| Interaction with microglia | Neuronal death due to release of proinflammatory cytokines. | [11][23][24][25] | |
| C1q released by microglia can induce A1 astrocytes. | [25] | ||
| Presence of complement receptors can increase phagocytosis. | [26][27][28] | ||
| Interaction with astrocytes | Neuronal death due to release of pro-inflammatory cytokines. | [25][29] | |
| Neurotoxic A1 astrocytes can activate the classical pathway and release pro-inflammatory cytokines. | [25][30][31][32] | ||
| NF-κB pathway activation via Aβ | Increased release of C3 via activation of NF-κB pathway resulting in microglia activation and release of pro-inflammatory cytokines. | [33][34] |
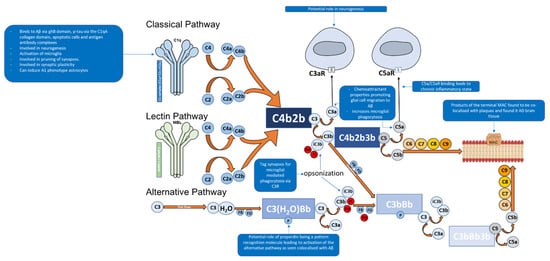
The C1 complex is composed of C1q (Figure 2), a charge pattern recognition molecule, and a tetramer of serine proteases C1r2 and C1s2 [11][6][12][13]. C1q serves as a molecular scaffold to C1r2 and C1s2; binding of C1q to a target ligand autoactivates C1r, which subsequently cleaves and activates C1s [12]. The activated C1s then goes on to cleave C4 to generate C4a and C4b which are an anaphylatoxin and opsonin, respectively [6][35]. C4b then recruits C2 [6]. C1s then cleaves C2 in to C2a and C2b [6]. The cleaved products C4b and C2b form C4b2b, a proconvertase complex [6][35][36]. The proconvertase complex is cleaved by C1s which results in C3 convertase, C4b2a [6][37]. C3 convertase cleaves multiple C3 proteins in to C3a and C3b [4][6]. C3b serves two functions, the first being an opsonin where it will covalently attach to the surface of a pathogens and drives the amplification of the complement alternative pathway, which results in opsonisation and phagocytosis of the tagged pathogen [4][6].The second function is where C3b leads to the downstream activation of the terminal pathway forming the MAC [4][6]. This is performed by C3b associating with C4b2b which results in the formation of C4b2b3b, known as C5 convertase of the classical pathway [6]. C5 convertase then cleaves C5 into C5a (an anaphylatoxin that also acts as a chemoattractant) and C5b, the latter facilitates the formation of the C5b, C6, C7, C8, and C9 complex (C5b-9), known as the MAC [38][4][6][39]. The MAC can then bind to the cell membrane and cause cell lysis [11].
The alternative pathway of the complement system is an essential amplification loop and the pathway is regulated by several control proteins, factors B, D, H, I, and properdin [40] (Figure 1). C3b molecules generated from any of the three pathways can covalently attach to the cell surface of a pathogen. C3b associates with factor B which results in the formation of C3bB, a Mg2+ dependent proconvertase complex [40]. Factor B is then cleaved by factor D to form Bb and Ba [41]. This results in the formation of C3bBb, which is the C3 convertase of the alternative pathway [40][41]. C3bBb cleaves C3 to C3a, an anaphylatoxin (which can also act as a chemoattractant) and C3b, of which C3b forms the alternative pathway amplification loop forming a new C3bBb complex each time [40][41].
The lectin pathway of the complement system is activated through pattern recognition molecules such as mannan-binding lectin (MBL) and ficolins, which bind to oligosaccharides on the surface of pathogens [66,67,68]. These pattern recognition molecules have an N-terminal collagenous region similar to C1q, but the C-terminal region differs as they contain C-type lectin domains [66,67,68]. Once activated, associated enzymes mannan-binding lectin serine protease 1 (MASP1) activates MASP2 which goes on to cleave C2 and C4 in to C2a, C2b, C4a and C4b; of which C2a and C4b form C3 convertase, C4b2a [66,67,68]. This pathway can then lead to the eventual formation of the MAC.
3. Complement System and Alzheimer’s Disease
3.1. Role of the Complement System in Central Nervous System Physiology
3.2. The Specific Role of the Complement System in Alzheimer’s Disease
4. Role of Glial Cells in AD and the Complement System
Astrocytes constitute approximately 30% of the cells in the CNS; they can morphologically be found in two forms, protoplasmic (in grey matter) and fibrous (in white matter) [5][49]. Astrocytes contribute to the BBB via their astrocytic end-feet, which line the surface of the brain and form a covering around the cerebral vessels and synapses [50][51]. Astrocytes also have a role in neuronal development and synaptogenesis, providing metabolic support to synapses [49][50]. Insults to the CNS causes astrocytes to change their morphology to become reactive astrocytes which exhibit hypertrophy of their processes and upregulate the release of glial fibrillary acidic protein (GFAP) and S100B; all of which are seen in AD brain tissue analysis [5][29]. Studies on AD human brains have identified reactive astrocytes in close proximity to Aβ plaques, and astrocytes containing Aβ plaques have also been stained positive; this may indicate a potential role of astrocytes in the clearance of Aβ in the early stages of AD [52][53]. Animal model studies have demonstrated that astrocytes migrate to Aβ via chemokines such as monocyte chemoattractant protein-1 (MCP-1), present in Aβ plaques and that astrocytes bind to and degrade Aβ [25]. Injection of Aβ oligomers induced a strong activation of astrocytes via the nuclear factor-kappa B (NF-κB) leading to the production of pro-inflammatory cytokines IL-1β and TNFα, surrounding injection sites and in close proximity to blood vessels which can draw in further glial cells, contributing to neuroinflammation [30] (Table 1). Furthermore, astrocytes have the ability to phagocytose Aβ [31][32]. Astrocytes exist in two reactivity states: A1 and A2 [33]. A2 astrocytes are neuroprotective as they perform homeostatic functions that helps restore activity of neurons and synapses after insults, whereas A1 astrocytes fail to perform this and convert to a neurotoxic form [33][34]. These neurotoxic A1 astrocytes can significantly increase activation of the complement classical pathway [33] (Table 1). Additionally, pro-inflammatory cytokines such as IL-1 and TNFα, and complement components including C1q, which are released by microglia, can also induce A1 astrocyte phenotype [33]. Extracellular tau aggregates can bind to astrocytes, get internalised via an integrin αV/β1 receptor; the integrin signalling pathway causes NF-κB activation leading to the release of several pro-inflammatory cytokines and chemokines that converts astrocytes to an A1-like neurotoxic state [34][54][55].
References
- World Health Organization Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 16 November 2020).
- Kumar, P.J.; Clark, M. Kumar & Clark’s Clinical Medicine, 7th ed.; Saunders Elsevier: Edinburgh, UK, 2011.
- Tenner, A.J. Complement-mediated events in Alzheimer’s disease: Mechanisms and potential therapeutic targets. J. Immunol. 2020, 204, 306–315.
- Walport, M.J. Advances in immunology: Complement (first of two parts). N. Engl. J. Med. 2001, 344, 1058–1066.
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediat. Inflamm. 2013, 2013, 342931.
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflamm. 2020, 17, 354.
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50.
- Coulthard, L.G.; Hawksworth, O.A.; Woodruff, T.M. Complement: The Emerging Architect of the Developing Brain. Trends Neurosci. 2018, 41, 373–384.
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178.
- Hirai, H.; Pang, Z.; Bao, D.; Miyazaki, T.; Li, L.; Miura, E.; Parris, J.; Rong, Y.; Watanabe, M.; Yuzaki, M.; et al. Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat. Neurosci. 2005, 8, 1534–1541.
- Bonifati, D.M.; Kishore, U. Role of complement in neurodegeneration and neuroinflammation. Mol. Immunol. 2007, 44, 999–1010.
- Lu, J.; Kishore, U. C1 complex: An adaptable proteolytic module for complement and non-complement functions. Front. Immunol. 2017, 8, 592.
- Mortensen, S.A.; Sander, B.; Jensen, R.K.; Pedersen, J.S.; Golas, M.M.; Jensenius, J.C.; Hansen, A.G.; Thiel, S.; Andersen, G.R. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc. Natl. Acad. Sci. USA 2017, 114, 986–991.
- Velazquez, P.; Cribbs, D.H.; Poulos, T.L.; Tenner, A.J. Aspartate residue 7 in amyloid β-protein is critical for classical complement pathway activation: Implications for Alzheimer’s disease pathogenesis. Nat. Med. 1997, 3, 77–79.
- Rogers, J.; Cooper, N.R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S.D.; Civin, W.H.; Brachova, L.; Bradt, B.; Ward, P.; et al. Complement activation by β-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10016–10020.
- Shen, Y.; Lue, L.F.; Yang, L.B.; Roher, A.; Kuo, Y.M.; Strohmeyer, R.; Goux, W.J.; Lee, V.; Johnson, G.V.W.; Webster, S.D.; et al. Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 2001, 305, 165–168.
- Wallis, R.; Dodds, A.W.; Mitchell, D.A.; Sim, R.B.; Reid, K.B.M.; Schwaeble, W.J. Molecular interactions between MASP-2, C4, and C2 and their activation fragments leading to complement activation via the lectin pathway. J. Biol. Chem. 2007, 282, 7844–7851.
- Héja, D.; Kocsis, A.; Dobó, J.; Szilágyi, K.; Szász, R.; Závodszky, P.; Pál, G.; Gál, P. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc. Natl. Acad. Sci. USA 2012, 109, 10498–10503.
- Gasque, P.; Chan, P.; Mauger, C.; Schouft, M.T.; Singhrao, S.; Dierich, M.P.; Morgan, B.P.; Fontaine, M. Identification and characterization of complement C3 receptors on human astrocytes. J. Immunol. 1996, 156, 2247–2255.
- Veerhuis, R.; Janssen, I.; De Groot, C.J.; Van Muiswinkel, F.L.; Hack, C.E.; Eikelenboom, P. Cytokines associated with amyloid plaques in Alzheimer’s disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp. Neurol. 1999, 160, 289–299.
- Rahpeymai, Y.; Hietala, M.A.; Wilhelmsson, U.; Fotheringham, A.; Davies, I.; Nilsson, A.K.; Zwirner, J.; Wetsel, R.A.; Gerard, C.; Pekny, M.; et al. Complement: A novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006, 25, 1364–1374.
- Ma, Y.; Ramachandran, A.; Ford, N.; Parada, I.; Prince, D.A. Remodeling of dendrites and spines in the C1q knockout model of genetic epilepsy. Epilepsia 2013, 54, 1232–1239.
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol. Brain Res. 1986, 1, 271–280.
- Ishiguro, K.; Shiratsuchi, A.; Sato, S.; Omori, A.; Arioka, M.; Kobayashi, S.; Uchida, T.; Imahori, K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993, 325, 167–172.
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457.
- Von Zahn, J.; Möller, T.; Kettenmann, H.; Nolte, C. Microglial phagocytosis is modulated by pro-and anti-inflammatory cytokines. NeuroReport 1997, 8, 3851–3856.
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705.
- Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Activated microglia desialylate their surface, stimulating complement receptor 3-mediated phagocytosis of neurons. Glia 2020, 68, 989–998.
- Beach, T.G.; McGeer, E.G. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res. 1988, 463, 357–361.
- Carrero, I.; Gonzalo, M.R.; Martin, B.; Sanz-Anquela, J.M.; Arevalo-Serrano, J.; Gonzalo-Ruiz, A. Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp. Neurol. 2012, 236, 215–227.
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209.
- Lee, S.J.; Seo, B.R.; Koh, J.Y. Metallothionein-3 modulates the amyloid beta endocytosis of astrocytes through its effects on actin polymerization. Mol. Brain 2015, 8, 84.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Wang, P.; Ye, Y. Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat. Commun. 2021, 12, 95.
- Gigli, I.; Fujita, T.; Nussenzweig, V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Proc. Natl. Acad. Sci. USA 1979, 76, 6596–6600.
- Krishnan, V.; Xu, Y.; Macon, K.; Volanakis, J.E.; Narayana, S.V.L. The structure of C2b, a fragment of complement component C2 produced during C3 convertase formation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 266–274.
- Ziccardi, R.J. Activation of the early components of the classical complement pathway under physiologic conditions. J. Immunol. 1981, 126, 1769–1773.
- Lo, M.W.; Woodruff, T.M. Complement: Bridging the innate and adaptive immune systems in sterile inflammation. J. Leukocyte Biol. 2020, 108, 339–351.
- McGeer, P.L.; Akiyama, H.; Itagaki, S.; McGeer, E.G. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci. Lett. 1989, 107, 341–346.
- Fromell, K.; Adler, A.; Åman, A.; Manivel, V.A.; Huang, S.; Dührkop, C.; Sandholm, K.; Ekdahl, K.N.; Nilsson, B. Assessment of the Role of C3(H2O) in the Alternative Pathway. Front. Immunol. 2020, 11, 530.
- Daha, M.R.; Fearon, D.T.; Austen, K.F. C3 requirements for formation of alternative pathway C5 convertase. J. Immunol. 1976, 117, 630–634.
- Benard, M.; Raoult, E.; Vaudry, D.; Leprince, J.; Falluel-Morel, A.; Gonzalez, B.J.; Galas, L.; Vaudry, H.; Fontaine, M. Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Mol. Immunol. 2008, 45, 3767–3774.
- Chu, Y.; Jin, X.; Parada, I.; Pesic, A.; Stevens, B.; Barres, B.; Prince, D.A. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7975–7980.
- Walker, D.G.; McGeer, P.L. Complement gene expression in human brain: Comparison between normal and Alzheimer disease cases. Mol. Brain Res. 1992, 14, 109–116.
- Kishore, U.; Gupta, S.K.; Perdikoulis, M.V.; Kojouharova, M.S.; Urban, B.C.; Reid, K.B.M. Modular organization of the carboxyl-terminal, globular head region of human C1q A, B, and C chains. J. Immunol. 2003, 171, 812–820.
- Yang, L.B.; Li, R.; Meri, S.; Rogers, J.; Shen, Y. Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer’s disease. J. Neurosci. 2000, 20, 7505–7509.
- Berg, A.; Zelano, J.; Stephan, A.; Thams, S.; Barres, B.A.; Pekny, M.; Pekna, M.; Cullheim, S. Reduced removal of synaptic terminals from axotomized spinal motoneurons in the absence of complement C3. Exp. Neurol. 2012, 237, 8–17.
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042.
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967.
- Eilert-Olsen, M.; Hjukse, J.B.; Thoren, A.E.; Tang, W.; Enger, R.; Jensen, V.; Pettersen, K.H.; Nagelhus, E.A. Astroglial endfeet exhibit distinct Ca2+ signals during hypoosmotic conditions. Glia 2019, 67, 2399–2409.
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530.
- Funato, H.; Yoshimura, M.; Yamazaki, T.; Saido, T.C.; Ito, Y.; Yokofujita, J.; Okeda, R.; Ihara, Y. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am. J. Pathol. 1998, 152, 983–992.
- Thal, D.R.; Schultz, C.; Dehghani, F.; Yamaguchi, H.; Braak, H.; Braak, E. Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol. 2000, 100, 608–617.
- McGeer, P.L.; McGeer, E.G. The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 1995, 21, 195–218.
- Johnstone, M.; Gearing, A.J.; Miller, K.M. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J. Neuroimmunol. 1999, 93, 182–193.
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318.
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553.
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458.
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. Activation of mGluR1 Mediates C1q-Dependent Microglial Phagocytosis of Glutamatergic Synapses in Alzheimer’s Rodent Models. Mol. Neurobiol. 2019, 56, 5568–5585.
- Husemann, J.; Loike, J.D.; Anankov, R.; Febbraio, M.; Silverstein, S.C. Scavenger receptors in neurobiology and neuropathology: Their role on microglia and other cells of the nervous system. Glia 2002, 40, 195–205.
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691.
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rube, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107.
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161.
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-β peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262.
- Fonseca, M.I.; Chu, S.H.; Hernandez, M.X.; Fang, M.J.; Modarresi, L.; Selvan, P.; MacGregor, G.R.; Tenner, A.J. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflamm. 2017, 14, 48.
- Gyorffy, B.A.; Kun, J.; Torok, G.; Bulyaki, E.; Borhegyi, Z.; Gulyassy, P.; Kis, V.; Szocsics, P.; Micsonai, A.; Matko, J.; et al. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc. Natl. Acad. Sci. USA 2018, 115, 6303–6308.
- Dejanovic, B.; Huntley, M.A.; De Mazière, A.; Meilandt, W.J.; Wu, T.; Srinivasan, K.; Jiang, Z.; Gandham, V.; Friedman, B.A.; Ngu, H.; et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 2018, 100, 1322–1336.e7.
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353.e5.


