Alzheimer’s disease is a type of dementia characterized by problems with short-term memory, cognition, and difficulties with activities of daily living. It is a progressive, neurodegenerative disorder. The complement system is an ancient part of the innate immune system and comprises of more than thirty serum and membrane-bound proteins. This system has three different activating pathways and culminates into the formation of a membrane attack complex that ultimately causes target cell lysis (usually pathogens) The complement system is involved in several important functions in the central nervous system (CNS) that include neurogenesis, synaptic pruning, apoptosis, and neuronal plasticity.
- neuroinflammation
- Alzheimer’s disease
- complement system
- microglia
1. Introduction
2. Role of the Complement System in CNS
The Complement System
Depending on the target ligand, the complement system can be activated by three pathways: classical, alternative, and lectin [11][5][12][15,16,44]. All three pathways share the common central component of the complement system C3, and downstream of C3 results in the formation of the MAC [11][15] (Figure 12). However, the pathways differ according to the ligand they can bind to [11][12][15,44].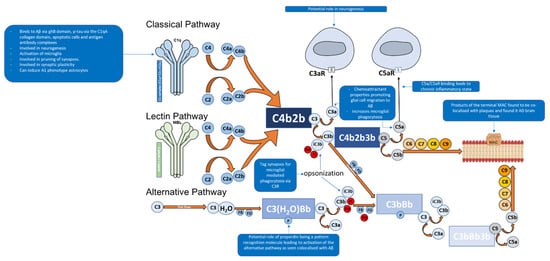
| Functions of Complement System | Role | Mechanism | Reference | ||
|---|---|---|---|---|---|
| Neuroprotection | Neurogenesis | Increased complement receptor activation in the development of cerebellar neurons in animal models. | [17] | [67] | |
| Disrupting C3aR signalling in mice models impairs neurogenesis. | [18] | [68] | |||
| CR2 is a negative regulator of neurogenesis. | [17][18] | [67,68] | |||
| Synaptic pruning | C1q | −/− | mice exhibit increased synaptic connections resulting in epilepsy, indicating an essential role in synaptic pruning. | [19][20] | [69,70] |
| Synaptic plasticity | C1q | −/− | mice show weak dendrites and spines. | [19] | [69] |
| Neuroinflammation | Binding with Aβ | Activation of classical pathway. | [9][10][21] | [42,43,71] | |
| Binding with Tau protein | Activation of complement system via classical pathway. | [12][22] | [44,72] | ||
| Interaction with microglia | Neuronal death due to release of proinflammatory cytokines. | [11][23][24][25] | [15,29,30,73] | ||
| C1q released by microglia can induce A1 astrocytes. | [25] | [73] | |||
| Presence of complement receptors can increase phagocytosis. | [26][27][28] | [74,75,76] | |||
| Interaction with astrocytes | Neuronal death due to release of pro-inflammatory cytokines. | [25][29] | [73,77] | ||
| Neurotoxic A1 astrocytes can activate the classical pathway and release pro-inflammatory cytokines. | [25][30][31][32] | [73,78,79,80] | |||
| NF-κB pathway activation via Aβ | Increased release of C3 via activation of NF-κB pathway resulting in microglia activation and release of pro-inflammatory cytokines. | [33][34] | [81,82] |
The C1 complex is composed of C1q (Figure 2), a charge pattern recognition molecule, and a tetramer of serine proteases C1r2 and C1s2 [11][6][12][13][15,40,44,45]. C1q serves as a molecular scaffold to C1r2 and C1s2; binding of C1q to a target ligand autoactivates C1r, which subsequently cleaves and activates C1s [12][44]. The activated C1s then goes on to cleave C4 to generate C4a and C4b which are an anaphylatoxin and opsonin, respectively [6][35][40,49]. C4b then recruits C2 [6][40]. C1s then cleaves C2 in to C2a and C2b [6][40]. The cleaved products C4b and C2b form C4b2b, a proconvertase complex [6][35][36][40,49,50]. The proconvertase complex is cleaved by C1s which results in C3 convertase, C4b2a [6][37][40,51]. C3 convertase cleaves multiple C3 proteins in to C3a and C3b [4][6][38,40]. C3b serves two functions, the first being an opsonin where it will covalently attach to the surface of a pathogens and drives the amplification of the complement alternative pathway, which results in opsonisation and phagocytosis of the tagged pathogen [4][6][38,40].The second function is where C3b leads to the downstream activation of the terminal pathway forming the MAC [4][6][38,40]. This is performed by C3b associating with C4b2b which results in the formation of C4b2b3b, known as C5 convertase of the classical pathway [6][40]. C5 convertase then cleaves C5 into C5a (an anaphylatoxin that also acts as a chemoattractant) and C5b, the latter facilitates the formation of the C5b, C6, C7, C8, and C9 complex (C5b-9), known as the MAC [38][4][6][39][37,38,40,52]. The MAC can then bind to the cell membrane and cause cell lysis [11][15].
The alternative pathway of the complement system is an essential amplification loop and the pathway is regulated by several control proteins, factors B, D, H, I, and properdin [40][53] (Figure 12). C3b molecules generated from any of the three pathways can covalently attach to the cell surface of a pathogen. C3b associates with factor B which results in the formation of C3bB, a Mg2+ dependent proconvertase complex [40][53]. Factor B is then cleaved by factor D to form Bb and Ba [41][54]. This results in the formation of C3bBb, which is the C3 convertase of the alternative pathway [40][41][53,54]. C3bBb cleaves C3 to C3a, an anaphylatoxin (which can also act as a chemoattractant) and C3b, of which C3b forms the alternative pathway amplification loop forming a new C3bBb complex each time [40][41][53,54].
The lectin pathway of the complement system is activated through pattern recognition molecules such as mannan-binding lectin (MBL) and ficolins, which bind to oligosaccharides on the surface of pathogens [66,67,68]. These pattern recognition molecules have an N-terminal collagenous region similar to C1q, but the C-terminal region differs as they contain C-type lectin domains [66,67,68]. Once activated, associated enzymes mannan-binding lectin serine protease 1 (MASP1) activates MASP2 which goes on to cleave C2 and C4 in to C2a, C2b, C4a and C4b; of which C2a and C4b form C3 convertase, C4b2a [66,67,68]. This pathway can then lead to the eventual formation of the MAC.
3. Complement System and Alzheimer’s Disease
3.1. Role of the Complement System in Central Nervous System Physiology
3.2. The Specific Role of the Complement System in Alzheimer’s Disease
The very important role of the complement system in AD pathophysiology is supported by neuropathology observed in vitro and in vivo. Studies have revealed that complement protein expression and complement activation lead to neuroinflammation, neuronal and synapse loss and subsequent neurodegeneration which is seen in AD patients. Complement proteins have been colocalised with Aβ plaques. A post-mortem study conducted by Rogers et al. (1992), who analysed AD patients brain tissues, showed elevated levels of C1q, C3, and C4 co-localisation with Aβ plaques compared with control samples [15][47] (Table 1). Another study observed elevated levels of C3 and C4 mRNA in the temporal cortex of AD brains [44][85]. A study by McGeer et al. (1989) identified an abundance of positive immunohistochemical staining of C1q, C3, and C4 and their colocalization with Aβ plaques and NFTs in AD brain tissue [39][52]. Specific staining for complement activation products such as C3b and the products of the terminal MAC (C5b-C9) in AD brain tissues has also been reported, indicating that that MAC can potentially cause neuronal loss and neurodegeneration in AD [44][85]. It is likely that the complement dysfunction may be contributing to neuroinflammation and subsequent neurodegeneration decades before clinical symptoms manifest in an individual with AD; this can be due to Aβ accumulation which overwhelms the complement system and drives the pathology of AD. In vitro studies have demonstrated that Aβ1-42 can directly activate the classical pathway by binding to C1q via its globular domain [45][86]. C1q can also bind to tau via the C1qA collagen domain and activate the classical pathway [46][87]. Thus, complement activation due to the binding of C1q to Aβ and tau can potentially contribute to neuroinflammation and neurodegeneration in AD. Animal models have allowed reproduction of the hallmarks of AD. Studies on C3 gene-deficient mice (C3−/−) that had a nerve injury showed a faster recovery compared with WT mice, indicating the complement system is involved in synapse removal and hinders recovery [47][88]. Another study examined the role of the complement on synapses in ageing mice and observed that C3−/− mice had better learning and memory in regions of the brain such as the hippocampus compared with their respective aged WT mice, suggesting that C3 or downstream complement components play a role in hampering synapses as a part of the aging process [48][89]. However, other studies have suggested that the complement system is essential for synaptic pruning [22][43][72,84] (Table 1). The complement system in a normal brain may aid in synaptic plasticity throughout life, but in the later stages of life, insults and accumulation of Aβ and NFTs can overactivate the complement or cause its dysfunction and fail to clear the hallmarks of AD 3.4. Role of Glial Cells in AD and the Complement System
Astrocytes constitute approximately 30% of the cells in the CNS; they can morphologically be found in two forms, protoplasmic (in grey matter) and fibrous (in white matter) [5][49][16,104]. Astrocytes contribute to the BBB via their astrocytic end-feet, which line the surface of the brain and form a covering around the cerebral vessels and synapses [50][51][105,106]. Astrocytes also have a role in neuronal development and synaptogenesis, providing metabolic support to synapses [49][50][104,105]. Insults to the CNS causes astrocytes to change their morphology to become reactive astrocytes which exhibit hypertrophy of their processes and upregulate the release of glial fibrillary acidic protein (GFAP) and S100B; all of which are seen in AD brain tissue analysis [5][29][16,77]. Studies on AD human brains have identified reactive astrocytes in close proximity to Aβ plaques, and astrocytes containing Aβ plaques have also been stained positive; this may indicate a potential role of astrocytes in the clearance of Aβ in the early stages of AD [52][53][107,108]. Animal model studies have demonstrated that astrocytes migrate to Aβ via chemokines such as monocyte chemoattractant protein-1 (MCP-1), present in Aβ plaques and that astrocytes bind to and degrade Aβ [25][73]. Injection of Aβ oligomers induced a strong activation of astrocytes via the nuclear factor-kappa B (NF-κB) leading to the production of pro-inflammatory cytokines IL-1β and TNFα, surrounding injection sites and in close proximity to blood vessels which can draw in further glial cells, contributing to neuroinflammation [30][78] (Table 1). Furthermore, astrocytes have the ability to phagocytose Aβ [31][32][79,80]. Astrocytes exist in two reactivity states: A1 and A2 [33][81]. A2 astrocytes are neuroprotective as they perform homeostatic functions that helps restore activity of neurons and synapses after insults, whereas A1 astrocytes fail to perform this and convert to a neurotoxic form [33][34][81,82]. These neurotoxic A1 astrocytes can significantly increase activation of the complement classical pathway [33][81] (Table 1). Additionally, pro-inflammatory cytokines such as IL-1 and TNFα, and complement components including C1q, which are released by microglia, can also induce A1 astrocyte phenotype [33][81]. Extracellular tau aggregates can bind to astrocytes, get internalised via an integrin αV/β1 receptor; the integrin signalling pathway causes NF-κB activation leading to the release of several pro-inflammatory cytokines and chemokines that converts astrocytes to an A1-like neurotoxic state [34][54][55][82,109,110].
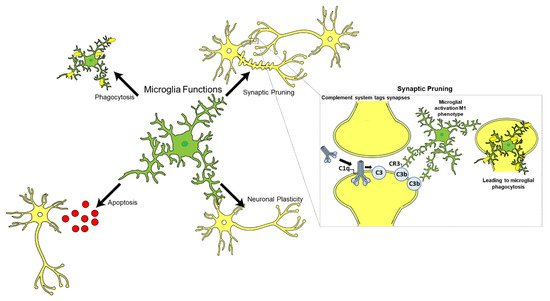
Post-mortem examination of AD brain tissues revealed an abundance of A1 astrocytes which also stained positive for C3 in brain regions affected by AD [33][81]. Furthermore, C1q was found to induce the A1 phenotype of astrocytes in vivo [33][81]. Following traumatic nerve crush injuries, C1q−/− mice showed a significant decrease in A1 astrocytes in comparison with WT controls [33][81], suggesting C1q influence on A1 astrocyte phenotype Microglia are the resident immune cells of the CNS and constitute approximately 10–15% of the total glial cell population in the adult human brain (Figure 23) [56][113]. Microglia play an essential role in immune surveillance in the CNS; they have long-ramified processes which they use to survey the microenvironment for cellular debris, pathogens, and misfolded proteins, and provide tropic support to the brain [57][58][114,115]. Other functions of microglia include neurogenesis, apoptosis, synaptic plasticity, and synaptic pruning in conjunction with the complement system, particularly C1q (Figure 23) [59][60][116,117].