 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stephani Joy Y. Macalino | + 3055 word(s) | 3055 | 2021-10-27 04:55:09 | | | |
| 2 | Peter Tang | Meta information modification | 3055 | 2021-12-29 07:00:57 | | |
Video Upload Options
Tuberculosis (TB) remains a serious threat to global public health, responsible for an estimated 1.5 million mortalities in 2018. Discovering new and more potent antibiotics that target novel TB protein targets is an attractive strategy towards controlling the global TB epidemic. In silico strategies can be applied at multiple stages of the drug discovery paradigm to expedite the identification of novel anti-TB therapeutics.
1. Introduction
2. Current Tuberculosis Management
2.1. Latent Tuberculosis Infection
2.2. Active Drug-Sensitive Tuberculosis
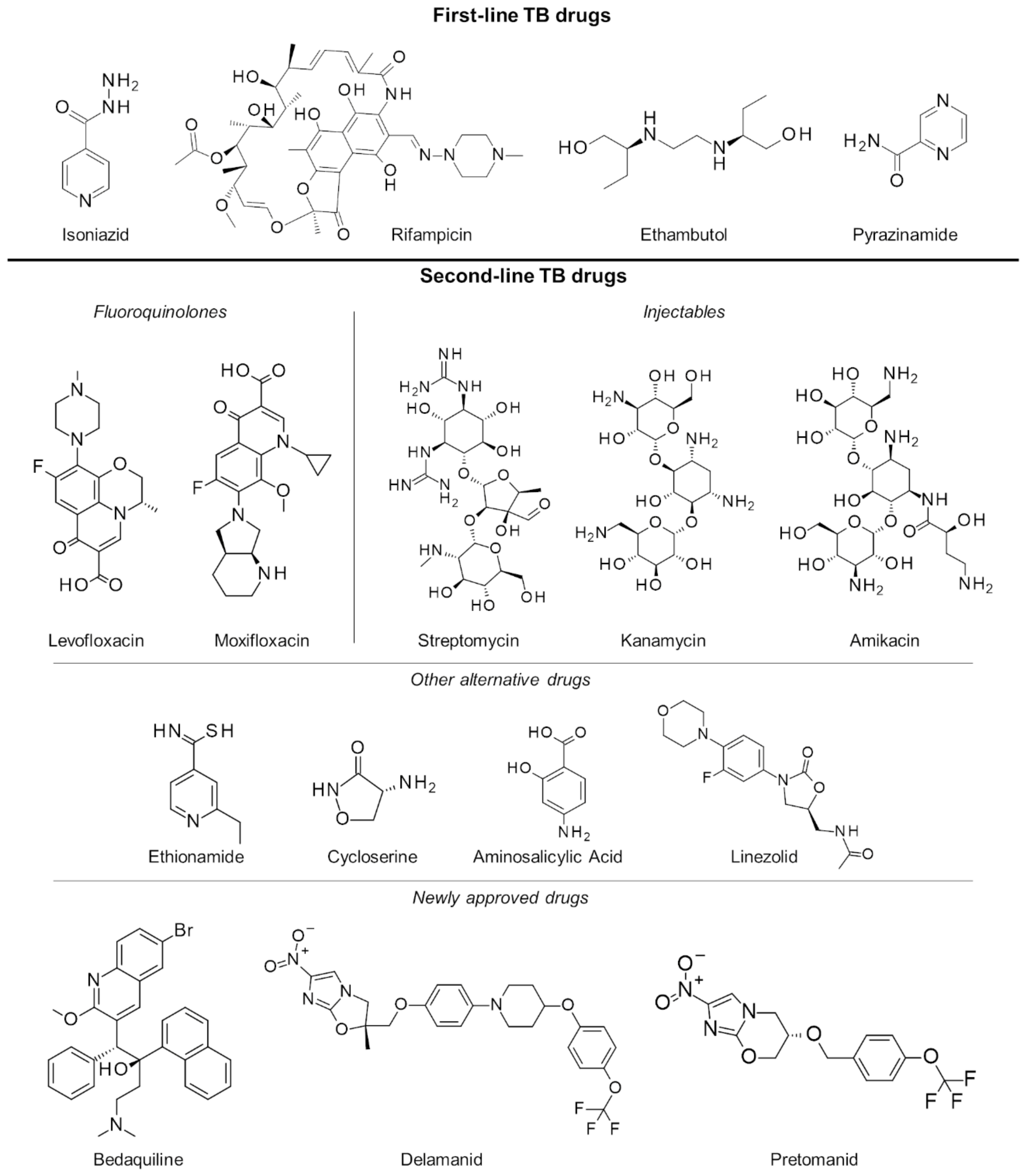
2.3. Multiple and Extensively Drug-Resistant Tuberculosis
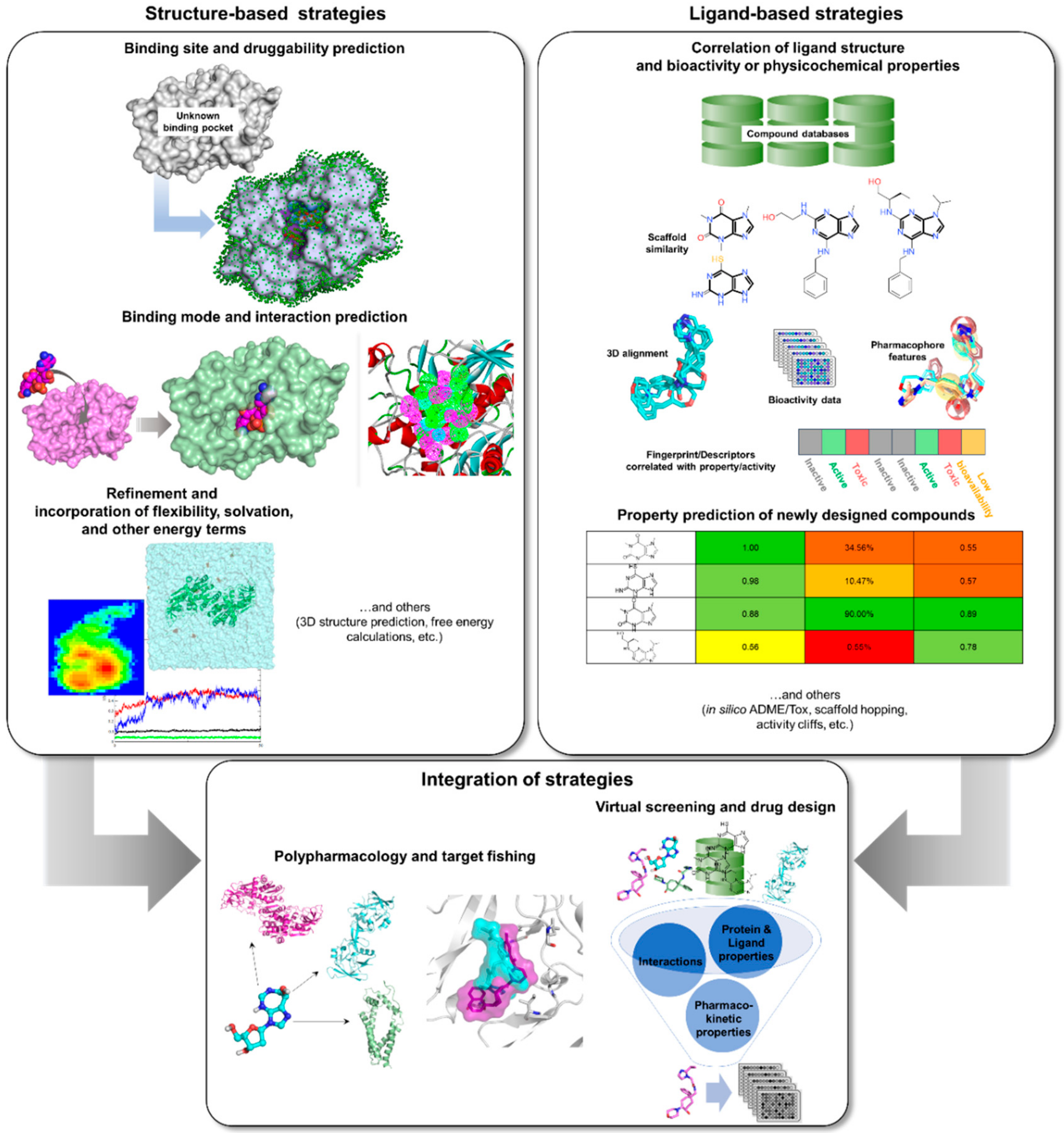
3. Rise of Computer-Aided Drug Design in TB Drug Discovery
|
Function |
Software/ Webserver Name |
Availability |
Website |
|---|---|---|---|
|
Comparative modeling |
SWISS-MODEL [32] |
Free webserver |
|
|
Structural geometry confirmation |
MODELLER [33] |
Free standalone program for academic license or commercially available through BIOVIA |
|
|
Robetta [34] |
Free webserver |
||
|
Prime [35] |
Commercially available through Schrödinger |
||
|
Free webserver or standalone program for academic license |
|||
|
Free webserver |
|||
|
Structural geometry confirmation |
PROCHECK [45] |
Free webserver and source code |
|
|
Druggability and binding site prediction Druggability and binding site prediction |
ProSA [46] |
Free webserver |
|
|
VERIFY3D [47] |
Free webserver |
||
|
ERRAT [48] |
Free webserver |
||
|
PockDrug [49] |
Free webserver |
http://pockdrug.rpbs.univ-paris-diderot.fr/cgi-bin/index.py?page=home |
|
|
DoGSiteScorer [50] |
Free webserver |
||
|
Free/open source platform |
|||
|
Free webserver |
|||
|
PocketQuery [56] |
Free webserver |
||
|
PASS [57] |
Free/open source platform |
||
|
SiteMap [58] |
Commercially available through Schrödinger |
||
|
Docking, pharmacophore, and virtual screening Docking, pharmacophore, and virtual screening |
ConCavity [59] |
Free webserver |
|
|
PrankWeb [60] |
Free webserver |
||
|
ProFunc [61] |
Free webserver |
||
|
Free standalone program |
|||
|
DOCK [64] |
Free/open source platform |
||
|
GOLD [65] |
Commercially available through CCDC |
https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ |
|
|
Glide [66] |
Commercially available through Schrödinger |
||
|
Induced Fit [67] |
Commercially available through Schrödinger |
||
|
FlexX [68] |
Commercially available through BioSolveIT |
||
|
RosettaLigand [69] |
Free/open source platform for academic license |
||
|
CDOCKER [70] |
Commercially available through BIOVIA |
||
|
Free webserver |
|||
|
Pharmer [73] |
Free/open source platform |
||
|
CATALYST [74] |
Commercially available through BIOVIA |
||
|
PharmGist [75] |
Free webserver |
||
|
LigandScout [76] |
Commercially available through Inte:Ligand |
||
|
SwissSimilarity [77] |
Free webserver |
||
|
LEA3D [78] |
Free webserver |
||
|
PyRx [79] |
Free (no support) or commercially available |
||
|
Phase [80] |
Commercially available through Schrödinger |
||
|
Molecular Dynamics |
Commercially available |
||
|
CHARMM [83] |
Free or commercially available through CHARMM or BIOVIA |
http://charmm.chemistry.harvard.edu/ https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/simulations.html |
|
|
CHARMMing [84] |
Free webserver |
||
|
Free/open source platform |
|||
|
NAMD [87] |
Free/open source platform |
||
|
Desmond [88] |
Commercially available through Schrödinger |
||
|
SwissParam [89] |
Free webserver |
||
|
CHARMM-GUI [90] |
Free webserver |
||
|
Free webserver |
|||
|
VMD [94] |
Free/open source platform |
||
|
Molecular Descriptors, Fingerprints, and Quantitative Structure-Activity Relationship |
Dragon [95] |
Commercially available through Talete |
|
|
E-Dragon [96] |
Free webserver |
||
|
Canvas [97] |
Commercially available through Schrödinger |
||
|
RDKit [98] |
Free/open source platform |
https://www.rdkit.org/docs/source/rdkit.ML.Descriptors.MoleculeDescriptors.html |
|
|
PyDescriptor [99] |
Free/open source platform |
||
|
Mordred [100] |
Free/open source platform |
||
|
Open3DQSAR [101] |
Free/open source platform |
||
|
ChemSAR [102] |
Free webserver |
||
|
SeeSAR [103] |
Commercially available through BioSolveIT |
||
|
Pharmacokinetic properties |
QikProp [104] |
Commercially available through Schrödinger |
|
|
ADMET Predictor [105] |
Commercially available through SimulationsPlus, Inc. |
||
|
ACD Percepta [106] |
Commercially available through ACD/Labs |
||
|
FAF-Drugs4 [107] |
Free webserver |
||
|
PatchSearch [108] |
Free webserver |
http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PatchSearch |
|
|
Commercially available through BIOVIA |
|||
|
PASS Online [111] |
Free webserver or commercially available standalone program |
||
|
SwissADME [112] |
Free webserver |
||
|
MetaSite [113] |
Commercially available through Molecular Discovery |
||
|
ToxPredict [114] |
Free webserver |
||
|
Free standalone software |
|||
|
Free webserver |
|||
|
Free webserver |
More available tools and detailed descriptions for the programs and servers can be found at https://www.click2drug.org/.
4. Edges and Pitfalls of In Silico Methods
References
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Primers 2016, 2, 16076.
- Havlir, D.V.; Getahun, H.; Sanne, I.; Nunn, P. Opportunities and challenges for HIV care in overlapping HIV and TB epidemics. JAMA 2008, 300, 423–430.
- Leung, C.C.; Yew, W.W.; Chan, C.K.; Chang, K.C.; Law, W.S.; Lee, S.N.; Tai, L.B.; Leung, E.C.; Au, R.K.; Huang, S.S.; et al. Smoking adversely affects treatment response, outcome and relapse in tuberculosis. Eur. Respir. J. 2015, 45, 738–745.
- Imtiaz, S.; Shield, K.D.; Roerecke, M.; Samokhvalov, A.V.; Lonnroth, K.; Rehm, J. Alcohol consumption as a risk factor for tuberculosis: Meta-analyses and burden of disease. Eur. Respir. J. 2017, 50, 1700216.
- Restrepo, B.I. Diabetes and Tuberculosis. In Understanding the Host Immune Response against Mycobacterium tuberculosis Infection; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–21.
- Getahun, H.; Matteelli, A.; Chaisson, R.E.; Raviglione, M. Latent Mycobacterium tuberculosis infection. N. Engl. J. Med. 2015, 372, 2127–2135.
- Miller, L.G.; Asch, S.M.; Yu, E.I.; Knowles, L.; Gelberg, L.; Davidson, P. A population-based survey of tuberculosis symptoms: How atypical are atypical presentations? Clin. Infect. Dis. 2000, 30, 293–299.
- World Health Organization (WHO). Global Tuberculosis Report 2019; World Health Organization (WHO): Geneva, Switzerland, 2019.
- World Health Organization (WHO). Guidelines on the Management of Latent Tuberculosis Infection; World Health Organization (WHO): Geneva, Switzerland, 2015.
- World Health Organization (WHO). Latent Tuberculosis infection: Updated and Consolidated Guidelines for Programmatic Management; World Health Organization (WHO): Geneva, Switzerland, 2018.
- Getahun, H.; Matteelli, A.; Abubakar, I.; Aziz, M.A.; Baddeley, A.; Barreira, D.; Den Boon, S.; Borroto Gutierrez, S.M.; Bruchfeld, J.; Burhan, E.; et al. Management of latent Mycobacterium tuberculosis infection: WHO guidelines for low tuberculosis burden countries. Eur. Respir. J. 2015, 46, 1563–1576.
- World Health Organization (WHO). Treatment of Tuberculosis: Guidelines; World Health Organization (WHO): Geneva, Switzerland, 2010.
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brozek, J.L.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, e147–e195.
- Volmink, J.; Garner, P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst. Rev. 2007, CD003343.
- Horsburgh, C.R., Jr.; Barry, C.E., 3rd; Lange, C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160.
- Saukkonen, J.J.; Cohn, D.L.; Jasmer, R.M.; Schenker, S.; Jereb, J.A.; Nolan, C.M.; Peloquin, C.A.; Gordin, F.M.; Nunes, D.; Strader, D.B.; et al. An official ATS statement: Hepatotoxicity of antituberculosis therapy. Am. J. Respir. Crit. Care Med. 2006, 174, 935–952.
- Dheda, K.; Barry, C.E., 3rd; Maartens, G. Tuberculosis. Lancet 2016, 387, 1211–1226.
- Dheda, K.; Gumbo, T.; Gandhi, N.R.; Murray, M.; Theron, G.; Udwadia, Z.; Migliori, G.B.; Warren, R. Global control of tuberculosis: From extensively drug-resistant to untreatable tuberculosis. Lancet. Respir. Med. 2014, 2, 321–338.
- World Health Organization (WHO). WHO Treatment Guidelines for Drug-Resistant Tuberculosis 2016 Update; World Health Organization (WHO): Geneva, Switzerland, 2016.
- Walker, J.; Tadena, N. J&J Tuberculosis Drug Gets Fast-Track Clearance. Wall St. J. 2013. Available online: https://www.wsj.com/articles/SB10001424127887323320404578213421059138236 (accessed on 27 November 2019).
- Mahajan, R. Bedaquiline: First FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res. 2013, 3, 1–2.
- European Medicines Agency (EMA). Deltyba Delamanid Summary of the European Public Assessment Report (EPAR) for Deltyba; EMA: Amsterdam, The Netherlands, 2014; pp. 1–3. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/deltyba (accessed on 15 November 2019).
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045.
- US FDA. FDA Approves New Drug for Treatment-Resistant Forms of Tuberculosis That Affects the Lungs; US FDA: Silver Spring, MD, USA, 2019.
- Baptista, R.; Fazakerley, D.M.; Beckmann, M.; Baillie, L.; Mur, L.A.J. Untargeted metabolomics reveals a new mode of action of pretomanid (PA-824). Sci. Rep. 2018, 8, 5084.
- Thompson, A.M.; Bonnet, M.; Lee, H.H.; Franzblau, S.G.; Wan, B.; Wong, G.S.; Cooper, C.B.; Denny, W.A. Antitubercular Nitroimidazoles Revisited: Synthesis and Activity of the Authentic 3-Nitro Isomer of Pretomanid. ACS Med. Chem. Lett. 2017, 8, 1275–1280.
- Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol. 2009, 2, 215–218.
- Reymond, J.-L.; van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 2010, 1, 30–38.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242.
- Ekins, S.; Freundlich, J.S.; Choi, I.; Sarker, M.; Talcott, C. Computational databases, pathway and cheminformatics tools for tuberculosis drug discovery. Trends Microbiol. 2011, 19, 65–74.
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701.
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385.
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2016, 86, 1–37.
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531.
- Schrödinger. Prime. Available online: https://www.schrodinger.com/prime (accessed on 26 October 2019).
- Zheng, W.; Zhang, C.; Bell, E.W.; Zhang, Y. I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Future Gener. Comput. Syst. 2019, 99, 73–85.
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008, 9, 40.
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181.
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8.
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738.
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins 2009, 77, 100–113.
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243.
- Hildebrand, A.; Remmert, M.; Biegert, A.; Soding, J. Fast and accurate automatic structure prediction with HHpred. Proteins 2009, 77, 128–132.
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248.
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291.
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410.
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404.
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519.
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.C. PockDrug-Server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015, 43, W436–W442.
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075.
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589.
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinf. 2009, 10, 168.
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367.
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003, 31, 3352–3355.
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118.
- Koes, D.R.; Camacho, C.J. PocketQuery: Protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res. 2012, 40, W387–W392.
- Brady, G.P., Jr.; Stouten, P.F. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des. 2000, 14, 383–401.
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389.
- Capra, J.A.; Laskowski, R.A.; Thornton, J.M.; Singh, M.; Funkhouser, T.A. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol. 2009, 5, e1000585.
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349.
- Laskowski, R.A.; Watson, J.D.; Thornton, J.M. ProFunc: A server for predicting protein function from 3D structure. Nucleic Acids Res. 2005, 33, W89–W93.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461.
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156.
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins 2003, 52, 609–623.
- Schrödinger. Glide. Available online: https://www.schrodinger.com/glide (accessed on 26 October 2019).
- Schrödinger. Induced Fit. Available online: https://www.schrodinger.com/induced-fit (accessed on 26 October 2019).
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489.
- Davis, I.W.; Baker, D. RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 2009, 385, 381–392.
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562.
- Bitencourt-Ferreira, G.; de Azevedo, W.F., Jr. Docking with SwissDock. Methods Mol. Biol. 2019, 2053, 189–202.
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277.
- Koes, D.R.; Camacho, C.J. Pharmer: Efficient and exact pharmacophore search. J. Chem. Inf. Model. 2011, 51, 1307–1314.
- Dassault Systèmes BIOVIA. Catalyst. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/pharmacophore-and-ligand-based-design.html (accessed on 26 October 2019).
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228.
- Inte:Ligand. LigandScout. Available online: http://www.inteligand.com/ligandscout/ (accessed on 26 October 2019).
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404.
- Douguet, D. e-LEA3D: A computational-aided drug design web server. Nucleic Acids Res. 2010, 38, W615–W621.
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250.
- Schrödinger. PHASE. Available online: https://www.schrodinger.com/phase (accessed on 26 October 2019).
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688.
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210.
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614.
- Miller, B.T.; Singh, R.P.; Klauda, J.B.; Hodoscek, M.; Brooks, B.R.; Woodcock, H.L., 3rd. CHARMMing: A new, flexible web portal for CHARMM. J. Chem. Inf. Model. 2008, 48, 1920–1929.
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25.
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802.
- Schrödinger. Desmond. Available online: https://www.schrodinger.com/desmond (accessed on 26 October 2019).
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368.
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865.
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690.
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154.
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–38.
- Helguera, A.M.; Combes, R.D.; Gonzalez, M.P.; Cordeiro, M.N. Applications of 2D descriptors in drug design: A DRAGON tale. Curr. Top. Med. Chem. 2008, 8, 1628–1655.
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463.
- Schrödinger. Canvas. Available online: https://www.schrodinger.com/canvas (accessed on 26 October 2019).
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 26 October 2019).
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18.
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4.
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2011, 17, 201–208.
- Dong, J.; Yao, Z.J.; Zhu, M.F.; Wang, N.N.; Lu, B.; Chen, A.F.; Lu, A.P.; Miao, H.; Zeng, W.B.; Cao, D.S. ChemSAR: An online pipelining platform for molecular SAR modeling. J. Cheminform. 2017, 9, 27.
- BioSolveIT. SeeSAR version 9.2. Available online: https://www.biosolveit.de/SeeSAR/ (accessed on 26 October 2019).
- Schrödinger. QikProp. Available online: https://www.schrodinger.com/qikprop (accessed on 26 October 2019).
- SimulationsPlus. ADMET Predictor. Available online: https://www.simulations-plus.com/software/admetpredictor/ (accessed on 26 October 2019).
- ACD/Labs. Percepta Platform. Available online: https://www.acdlabs.com/products/percepta/ (accessed on 26 October 2019).
- Miteva, M.A.; Violas, S.; Montes, M.; Gomez, D.; Tuffery, P.; Villoutreix, B.O. FAF-Drugs: Free ADME/tox filtering of compound collections. Nucleic Acids Res. 2006, 34, W738–W744.
- Rasolohery, I.; Moroy, G.; Guyon, F. PatchSearch: A Fast Computational Method for Off-Target Detection. J. Chem. Inf. Model. 2017, 57, 769–777.
- Dassault Systèmes BIOVIA. DS TOPKAT. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 26 October 2019).
- Dassault Systèmes BIOVIA. DS ADMET. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-pipeline-pilot/component-collections/adme-tox.html (accessed on 26 October 2019).
- Poroikov, V.; Filimonov, D.; Lagunin, A.; Gloriozova, T.; Zakharov, A. PASS: Identification of probable targets and mechanisms of toxicity. SAR QSAR Environ. Res. 2007, 18, 101–110.
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717.
- Cruciani, G.; Carosati, E.; De Boeck, B.; Ethirajulu, K.; Mackie, C.; Howe, T.; Vianello, R. MetaSite: Understanding metabolism in human cytochromes from the perspective of the chemist. J. Med. Chem. 2005, 48, 6970–6979.
- Tcheremenskaia, O.; Benigni, R.; Nikolova, I.; Jeliazkova, N.; Escher, S.E.; Batke, M.; Baier, T.; Poroikov, V.; Lagunin, A.; Rautenberg, M.; et al. OpenTox predictive toxicology framework: Toxicological ontology and semantic media wiki-based OpenToxipedia. J. Biomed. Semant. 2012, 3, S7.
- Smiesko, M.; Vedani, A. VirtualToxLab: Exploring the Toxic Potential of Rejuvenating Substances Found in Traditional Medicines. Methods Mol. Biol. 2016, 1425, 121–137.
- Vedani, A.; Dobler, M.; Smiesko, M. VirtualToxLab—A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 2012, 261, 142–153.
- Vedani, A.; Smiesko, M.; Spreafico, M.; Peristera, O.; Dobler, M. VirtualToxLab—In silico prediction of the toxic (endocrine-disrupting) potential of drugs, chemicals and natural products. Two years and 2000 compounds of experience: A progress report. ALTEX 2009, 26, 167–176.
- Vedani, A.; Dobler, M.; Spreafico, M.; Peristera, O.; Smiesko, M. VirtualToxLab—In silico prediction of the toxic potential of drugs and environmental chemicals: Evaluation status and internet access protocol. ALTEX 2007, 24, 153–161.
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. Correction to “admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties”. J. Chem. Inf. Model. 2019.
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069.
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105.
- Rudik, A.; Bezhentsev, V.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. Metatox-Web application for generation of metabolic pathways and toxicity estimation. J. Bioinform. Comput. Biol. 2019, 17, 1940001.
- Rudik, A.V.; Bezhentsev, V.M.; Dmitriev, A.V.; Druzhilovskiy, D.S.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. MetaTox: Web Application for Predicting Structure and Toxicity of Xenobiotics’ Metabolites. J. Chem. Inf. Model. 2017, 57, 638–642.
- Villoutreix, B.O.; Eudes, R.; Miteva, M.A. Structure-based virtual ligand screening: Recent success stories. Comb. Chem. High Throughput Screen. 2009, 12, 1000–1016.
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141.
- Clark, D.E. What has virtual screening ever done for drug discovery? Expert Opin. Drug Discov. 2008, 3, 841–851.
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martinez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881.
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provaznik, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581.
- Coupez, B.; Lewis, R.A. Docking and scoring—Theoretically easy, practically impossible? Curr. Med. Chem. 2006, 13, 2995–3003.
- Geppert, H.; Vogt, M.; Bajorath, J. Current trends in ligand-based virtual screening: Molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 2010, 50, 205–216.
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 2008, 22, 133–139.
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Shaw, D.E. Picosecond to Millisecond Structural Dynamics in Human Ubiquitin. J. Phys. Chem. B 2016, 120, 8313–8320.
- Noe, F. Beating the millisecond barrier in molecular dynamics simulations. Biophys. J. 2015, 108, 228–229.
- Shi, J.; Nobrega, R.P.; Schwantes, C.; Kathuria, S.V.; Bilsel, O.; Matthews, C.R.; Lane, T.J.; Pande, V.S. Atomistic structural ensemble refinement reveals non-native structure stabilizes a sub-millisecond folding intermediate of CheY. Sci. Rep. 2017, 7, 44116.
- Fujita, T. Recent Success Stories Leading to Commercializable Bioactive Compounds with the Aid of Traditional QSAR Procedures. QSAR 1997, 16, 107–112.
- Gao, Q.; Yang, L.; Zhu, Y. Pharmacophore based drug design approach as a practical process in drug discovery. Curr. Comput. Aided Drug Des. 2010, 6, 37–49.
- Sardari, S.; Dezfulian, M. Cheminformatics in anti-infective agents discovery. Mini Rev. Med. Chem. 2007, 7, 181–189.
- Bostrom, J.; Norrby, P.O.; Liljefors, T. Conformational energy penalties of protein-bound ligands. J. Comput. Aided Mol. Des. 1998, 12, 383–396.
- Perola, E.; Charifson, P.S. Conformational analysis of drug-like molecules bound to proteins: An extensive study of ligand reorganization upon binding. J. Med. Chem. 2004, 47, 2499–2510.
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design—A review. Curr. Top. Med. Chem. 2010, 10, 95–115.
- Hu, Y.; Bajorath, J. Extending the activity cliff concept: Structural categorization of activity cliffs and systematic identification of different types of cliffs in the ChEMBL database. J. Chem. Inf. Model. 2012, 52, 1806–1811.
- Hu, Y.; Stumpfe, D.; Bajorath, J. Advancing the activity cliff concept. F1000Res 2013, 2, 199.
- Stumpfe, D.; de la Vega de Leon, A.; Dimova, D.; Bajorath, J. Advancing the activity cliff concept, part II. F1000Res 2014, 3, 75.

