Tuberculosis (TB) remains a serious threat to global public health, responsible for an estimated 1.5 million mortalities in 2018. Discovering new and more potent antibiotics that target novel TB protein targets is an attractive strategy towards controlling the global TB epidemic. In silico strategies can be applied at multiple stages of the drug discovery paradigm to expedite the identification of novel anti-TB therapeutics.
- tuberculosis
- druggability
- docking
- pharmacophore
- MD simulation
- QSAR
- DFT
1. Introduction
2. Current Tuberculosis Management
2.1. Latent Tuberculosis Infection
2.2. Active Drug-Sensitive Tuberculosis
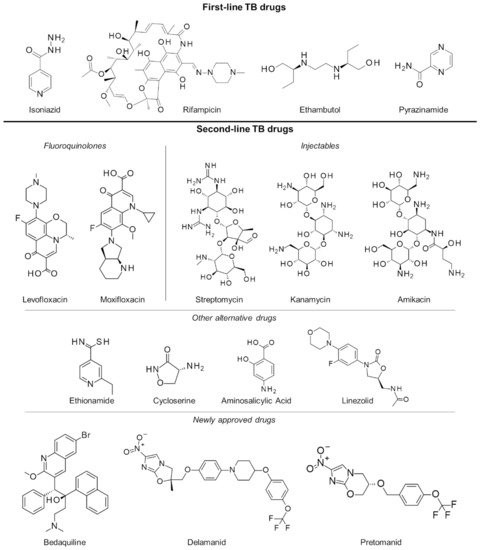
2.3. Multiple and Extensively Drug-Resistant Tuberculosis
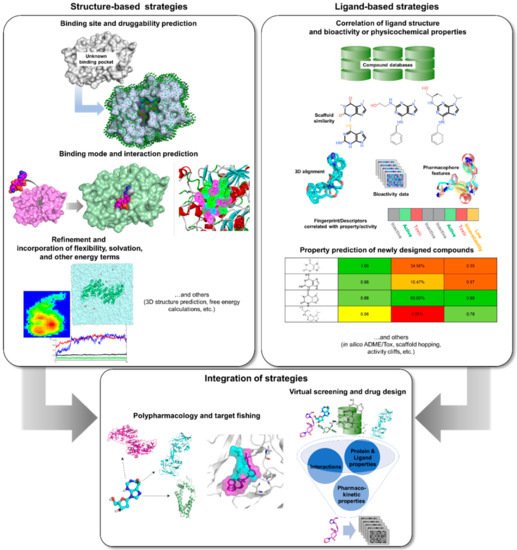
3. Rise of Computer-Aided Drug Design in TB Drug Discovery
|
Function |
Software/ Webserver Name |
Availability |
Website |
|---|---|---|---|
|
Comparative modeling |
SWISS-MODEL [32] |
Free webserver |
|
|
Structural geometry confirmation |
MODELLER [33] |
Free standalone program for academic license or commercially available through BIOVIA |
|
|
Robetta [34] |
Free webserver |
||
|
Prime [35] |
Commercially available through Schrödinger |
||
|
Free webserver or standalone program for academic license |
|||
|
Free webserver |
|||
|
Structural geometry confirmation |
PROCHECK [45] |
Free webserver and source code |
|
|
Druggability and binding site prediction Druggability and binding site prediction |
ProSA [46] |
Free webserver |
|
|
VERIFY3D [47] |
Free webserver |
||
|
ERRAT [48] |
Free webserver |
||
|
PockDrug [49] |
Free webserver |
http://pockdrug.rpbs.univ-paris-diderot.fr/cgi-bin/index.py?page=home |
|
|
DoGSiteScorer [50] |
Free webserver |
||
|
Free/open source platform |
|||
|
Free webserver |
|||
|
PocketQuery [56] |
Free webserver |
||
|
PASS [57] |
Free/open source platform |
||
|
SiteMap [58] |
Commercially available through Schrödinger |
||
|
Docking, pharmacophore, and virtual screening Docking, pharmacophore, and virtual screening |
ConCavity [59] |
Free webserver |
|
|
PrankWeb [60] |
Free webserver |
||
|
ProFunc [61] |
Free webserver |
||
|
Free standalone program |
|||
|
DOCK [64] |
Free/open source platform |
||
|
GOLD [65] |
Commercially available through CCDC |
https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ |
|
|
Glide [66] |
Commercially available through Schrödinger |
||
|
Induced Fit [67] |
Commercially available through Schrödinger |
||
|
FlexX [68] |
Commercially available through BioSolveIT |
||
|
RosettaLigand [69] |
Free/open source platform for academic license |
||
|
CDOCKER [70] |
Commercially available through BIOVIA |
||
|
Free webserver |
|||
|
Pharmer [73] |
Free/open source platform |
||
|
CATALYST [74] |
Commercially available through BIOVIA |
||
|
PharmGist [75] |
Free webserver |
||
|
LigandScout [76] |
Commercially available through Inte:Ligand |
||
|
SwissSimilarity [77] |
Free webserver |
||
|
LEA3D [78] |
Free webserver |
||
|
PyRx [79] |
Free (no support) or commercially available |
||
|
Phase [80] |
Commercially available through Schrödinger |
||
|
Molecular Dynamics |
Commercially available |
||
|
CHARMM [83] |
Free or commercially available through CHARMM or BIOVIA |
http://charmm.chemistry.harvard.edu/ https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/simulations.html |
|
|
CHARMMing [84] |
Free webserver |
||
|
Free/open source platform |
|||
|
NAMD [87] |
Free/open source platform |
||
|
Desmond [88] |
Commercially available through Schrödinger |
||
|
SwissParam [89] |
Free webserver |
||
|
CHARMM-GUI [90] |
Free webserver |
||
|
Free webserver |
|||
|
VMD [94] |
Free/open source platform |
||
|
Molecular Descriptors, Fingerprints, and Quantitative Structure-Activity Relationship |
Dragon [95] |
Commercially available through Talete |
|
|
E-Dragon [96] |
Free webserver |
||
|
Canvas [97] |
Commercially available through Schrödinger |
||
|
RDKit [98] |
Free/open source platform |
https://www.rdkit.org/docs/source/rdkit.ML.Descriptors.MoleculeDescriptors.html |
|
|
PyDescriptor [99] |
Free/open source platform |
||
|
Mordred [100] |
Free/open source platform |
||
|
Open3DQSAR [101] |
Free/open source platform |
||
|
ChemSAR [102] |
Free webserver |
||
|
SeeSAR [103] |
Commercially available through BioSolveIT |
||
|
Pharmacokinetic properties |
QikProp [104] |
Commercially available through Schrödinger |
|
|
ADMET Predictor [105] |
Commercially available through SimulationsPlus, Inc. |
||
|
ACD Percepta [106] |
Commercially available through ACD/Labs |
||
|
FAF-Drugs4 [107] |
Free webserver |
||
|
PatchSearch [108] |
Free webserver |
http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PatchSearch |
|
|
Commercially available through BIOVIA |
|||
|
PASS Online [111] |
Free webserver or commercially available standalone program |
||
|
SwissADME [112] |
Free webserver |
||
|
MetaSite [113] |
Commercially available through Molecular Discovery |
||
|
ToxPredict [114] |
Free webserver |
||
|
Free standalone software |
|||
|
Free webserver |
|||
|
Free webserver |
More available tools and detailed descriptions for the programs and servers can be found at https://www.click2drug.org/.
4. Edges and Pitfalls of In Silico Methods
This entry is adapted from the peer-reviewed paper 10.3390/molecules25030665