Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yu-Tzu Tai | + 2329 word(s) | 2329 | 2021-12-15 03:25:38 | | | |
| 2 | Yvaine Wei | Meta information modification | 2329 | 2021-12-27 01:51:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tai, Y. New Frontier of Targeted Immunotherapy in Multiple Myeloma. Encyclopedia. Available online: https://encyclopedia.pub/entry/17559 (accessed on 26 June 2026).
Tai Y. New Frontier of Targeted Immunotherapy in Multiple Myeloma. Encyclopedia. Available at: https://encyclopedia.pub/entry/17559. Accessed June 26, 2026.
Tai, Yu-Tzu. "New Frontier of Targeted Immunotherapy in Multiple Myeloma" Encyclopedia, https://encyclopedia.pub/entry/17559 (accessed June 26, 2026).
Tai, Y. (2021, December 25). New Frontier of Targeted Immunotherapy in Multiple Myeloma. In Encyclopedia. https://encyclopedia.pub/entry/17559
Tai, Yu-Tzu. "New Frontier of Targeted Immunotherapy in Multiple Myeloma." Encyclopedia. Web. 25 December, 2021.
Copy Citation
The approval of monoclonal antibody (MoAb) against CD38 (daratumumab) and SLAMF7 (elotuzumab) in relapsed and refractory MM (RRMM) represents an important milestone in the development of targeted immunotherapy in MM. These MoAb-based agents significantly induce cytotoxicity of MM cells via multiple effector-dependent mechanisms and can further induce immunomodulation to repair a dysfunctional tumor immune microenvironment.
multiple myeloma
MM
immunotherapy
immunomodulatory drugs
IMiDs
MoAb
CD38
SLAMF7
BCMA
bone marrow (BM) microenvironment
GPRC5D
1. Introduction
The development and introduction of the proteasome inhibitor (PI) bortezomib and immunomodulatory drugs (IMiDs), including thalidomide and lenalidomide, has revolutionized the treatment paradigm for multiple myeloma (MM). Second-generation drugs within the same classes, such as carfilzomib and ixazomib (PIs) and pomalidomide (IMiDs), further improve the response rate, survival, and safety profile [1][2][3]. The incorporation of autologous stem cell transplantation in eligible patients has also prolonged survival with more durable disease control [4][5]. However, disease recurrence remains common for most MM patients. Since drug-resistant clones constantly emerge and evolve, leading to a low 5-year overall survival rate in real-world data [6]. The clinical outcomes of patients with relapsed or refractory MM (RRMM) are dismally poor because of the gradually decreased durability of the response to successive lines of anti-MM therapy [7].
Accumulating studies for the past decades have defined that the bone marrow (BM) microenvironment is essential in supporting MM cell growth, survival, and drug resistance. MM cells are in close contact with surrounding BM accessory cells through bi-directional interactions, including stromal cells (BMSCs) [8], osteoclasts (OCs) [9][10], regulatory T (Treg) or B (Breg) cells [11][12][13], myeloid-derived suppressor cells (MDSCs) [14], tumor-associated macrophages (TAMs) [15], and plasmacytoid dendritic cells [16]. These non-MM cells, in turn, secrete abnormal levels of a variety of cytokines and growth factors in a paracrine fashion to promote pathogenesis of MM, including interleukin-6 (IL-6), IL-10, MIP-1α/β, transforming growth factor-beta (TGFβ), stromal cell-derived factor-1 (SDF-1), and a proliferation-inducing ligand (APRIL) [9][17][18][19]. Furthermore, changes in BM accessory cells and cytokines, either secreted by accessory cells or MM cells via autocrine or paracrine manners, contribute to myeloma cell immune escape, inhibition of myeloma-specific T effector cells, induction of T-cell anergy, and abnormality in Treg cells, resulting in an immunosuppressive microenvironment that impairs immunotherapy [20].
Monoclonal antibodies (MoAbs) binding to selective molecules on the surface of cancer cells have transformed cancer treatment. In principle, these biologically based molecules/proteins induce tumor cell killing mainly dependent on effector functions, including antibody-dependent cellular cytotoxicity (ADCC) via CD16-expressing effector cells (i.e., NK cells, neutrophils, monocytes), complement-dependent cytotoxicity (CDC), and/or antibody-dependent cellular phagocytosis (ADCP) via macrophages. These primary mechanisms of action are distinct from small molecules used in conventional chemotherapies, which directly induce tumor cell apoptosis and are largely independent of immune effector function. The first two therapeutic MoAbs available for RRMM patients are MoAbs targeting CD38 (daratumumab) and SLAMF7 (also named CS1) (elotuzumab), approved by the U.S. Food and Drug Administration (FDA) in 2015 [21][22]. These represent an important breakthrough for effective targeted immune-based therapies in MM. Importantly, results obtained from preclinical and clinical studies of both MoAbs thus far have shown that these first-generation targeting bio-molecules also affect the immunosuppressive non-MM cell components in addition to MM cells [11][12][13][23][24][25]. These findings have inspired many investigations on identifying the patho-immunological roles of various immune regulatory cell subsets and molecules regulating their function using in vitro, ex vivo, and in vivo models.
2. Pathophysiological Function for Validated MM Target Antigens and Their Related Immunotherapies
The anti-MM mechanisms of targeted immunotherapeutic bio-reagents are mainly derived from the MoAbs, which detect and engage with selective proteins on MM cell membrane followed by the induction of NK cell-mediated killing (Figure 1 and Figure 2). These target antigens are chosen based on their differential expression and/or critical roles in growth, survival, and drug resistance in MM cells.
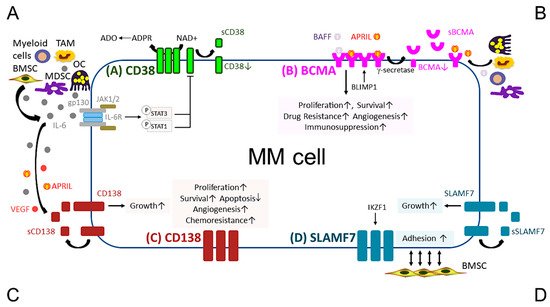
Figure 1. Pathobiological function of validated MM target antigens commonly used in current MM immunotherapies. (A) CD38, a receptor and an ectoenzyme, regulates the conversion of NAD+ to ADO, which contributes to the immunocompromised tumor microenvironment. IL-6, an important MM growth, survival, and immunosuppressive cytokine, mainly produced by non-myeloma BM cells (i.e., BMSCs, MDSC, OC, and TAM), binds to IL-6R, which interacts with gp130 to activate the JAK1/2-STAT1/3 signaling pathway and subsequently downregulates CD38 expression in MM cells. (B) BCMA is the cognate receptor for APRIL and BAFF, which are present in the BM microenvironment. APRIL, an important plasma cell factor, preferentially binds to BCMA when compared with BAFF, to induce signaling pathways critical to promote survival, proliferation, and drug resistance, as well as immunosuppression of MM cells. BCMA expression is induced by BLIMP1, a key plasma cell transcriptional regulator, and BCMA protein is cleaved by gamma (γ)-secretase, resulting in the soluble form (sBCMA) that is detected in MM patient serum and correlated with disease advancement. (C) CD138, the transmembrane heparan sulfate proteoglycan syndecan-1, is overexpressed in malignant plasma cells and its levels are associated with increased proliferation and survival, as well as decreased apoptosis in MM cell lines and patient MM cells. Levels of shed CD138 (sCD138) cleaved by metalloproteinases and heparanase in MM patient serum samples are also linked to disease progression and sCD138 acts locally or distally with various effector molecules (i.e., IL-6, APRIL, and VEGF) to impact tumor progression. (D) SLAMF7, also named CS1, promotes adhesion of MM cells to BMSCs. The expression of SLAMF7 is regulated by the transcriptional factor IKZF1 (Ikaros), a MM-related target by lenalidomide and pomalidomide, in MM cells. Soluble SLAMF7 (sSLAMF7) is detected in patient serum and promotes MM cell growth via homophilic interaction with SLAMF7 on MM cells. Among these four MM antigens, BCMA has the most limited expression at the transcript and protein levels in plasma cells but no other normal tissues except a minute subset of plasmacytoid dendritic cell. ADO, adenosine; ADPR, ADP-ribose; APRIL, A proliferation inducing ligand; BAFF, B-cell activating factor; BMSC, bone marrow stromal cell; JAK, Janus kinase; MDSC, myeloid-derived suppressor cell; NAD+, nicotinamide adenine dinucleotide; OC, osteoclast; TAM, tumor-associated macrophages; VEGF, vascular endothelial growth factor.
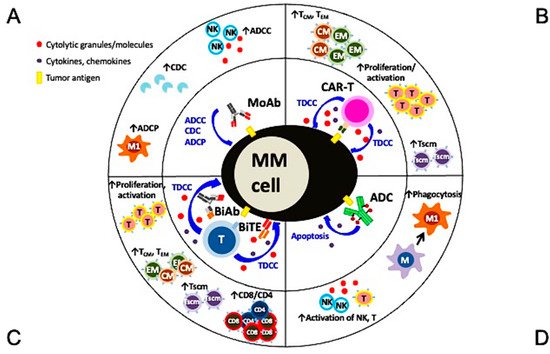
Figure 2. Multiple mechanisms of action and immunomodulatory effects of current targeted immunotherapies in MM. (A) Monoclonal antibodies (MoAbs) identify and bind to specific antigens to trigger MM cell lysis via multiple immune-dependent mechanisms, including ADCC, ADCP, and CDC. (B) The single chain fragment variant of the CAR-T cell first targets the MM-specific antigen and potent MM cell lysis is induced in a major histocompatibility complex-independent manner followed by increased proliferation and activation of T effector cells, including those with central and effector memory phenotypes. (C) BiAb or BiTE, off-shelf products different from autologous CAR-T cells, simultaneously binds to specific tumor antigen on MM cells and CD3 on T cells to trigger potent MM cell killing as well as proliferation of immune effector T cells. T cells with memory phenotypes (Tcm, Tem, Tscm) and the major cytolytic T cells (CD8+ cells) are increased significantly. (D) The MoAb portion of antibody drug conjugate (ADC) binds to tumor antigen on MM cell membrane and the ADC is endocytosed followed by the release of potent cytotoxic payloads from lysosomes to directly induce MM cell apoptosis. Payloads used in ADC are typically even more potent than those used in chemotherapies to improve superior specific killing of tumors cells but not the surrounding normal tissues. Depending on the design of the ADC, NK cells and macrophages are directly or indirectly activated to induce ADCC and phagocytosis, respectively, to further augment killing of tumor cells. ADCC, antibody-dependent cellular cytotoxicity (NK cells are the predominant effector cells. Other CD16-expressing effectors including monocytes and neutrophils are also capable to induce ADCC); ADCP, antibody-dependent cellular phagocytosis (macrophage (M), especially with the M1 characteristics, are the key effector cells whereas macrophages with M2 features (TAM in Figure 1) promote tumor growth); BiAb, bispecific antibody engaging with T cells; BiTE, bispecific T cell engager; CDC, complement-dependent cytotoxicity; Tcm, central memory T cell; Tem, effector memory T cell; Tscm, stem cell-like memory T cell.
2.1. CD38
CD38, a type II transmembrane glycoprotein, was first identified as a marker of cell activation and proliferation in lymphocytes. It regulates cell migration [26] and receptor-mediated adhesion via interaction with endothelial CD31 or hyaluronic acid [27]. CD38 receptor also exhibits ecto-enzymatic activity, involved in the metabolism of cytoplasmic nicotinamide adenine dinucleotide phosphate (NADP) and extracellular nicotinamide adenine dinucleotide (NAD+) [28]. In addition, CD38 interacts with its substrate, NAD+, to increase production of Ca2+ mobilizing compounds; i.e., cyclic adenosine diphosphate ribose (CADPR), ADP ribose (ADPR), and nicotinic acid adenine dinucleotide phosphate [29][30][31]. ADPR is further converted to adenosine monophosphate (AMP) by CD203a and then adenosine (ADO), which exhibits immunosuppressive activity via a reduction in immune cell activity and induction of differentiation of osteoclast, one of the most immunosuppressive BM accessory cells [32][33][34][35]. Moreover, the malignant plasma cells further use aerobic glycolysis to promote an acidic BM, which together with CD38 highly expressed on their surface induce the generation of AMP and ADO.
2.2. SLAMF7 (CS1)
SLAMF7 is a member of the immunoglobulin gene superfamily (signaling lymphocyte activation molecule family) and associated with cytotoxic effector, humoral and auto-immunity, and cell survival/adhesion, as well as lymphocyte development [36][37]. SLAMF7 is also expressed on the surface of certain subsets of immune cells, including NK cells, cytotoxic T lymphocytes (CD8+ cells), B lymphocytes, and mature dendritic cells. SLAMF7 itself can serve as a receptor of NK cell and it is a self-ligand that exhibits a homophilic interaction to augment NK cell-mediated cytotoxicity [38]. Moreover, SLAMF7 on the surface of B cells is upregulated during B cell activation to promote proliferation of naive and memory B cells and cytokine production [39].
2.3. CD138 (Syndecan-1)
CD138 (syndecan 1), a member of the syndecan family of type I transmembrane proteoglycan, has been commonly used as a prognostic marker in MM, since its expression level is elevated in malignant versus normal plasma cells [40]. CD138 modulates various biological processes, including proliferation [41], adhesion [42], migration [43], endocytosis [44], macropinocytosis [45], immunomodulation [46], and regulation of heparan sulfate proteoglycans [47]. Increased CD38 expression promotes proliferation and survival of MM cells, as well as angiogenesis and IL-6 receptor sensitivity in MM cells [48][49]. IL-6-induced growth and survival signaling cascades upon binding to IL-6R is further augmented in MM cells overexpressing CD138, indicating cross-talks between CD138 and IL6R in the progression of MM. Importantly, high CD138 expression is linked to enhanced malignant plasma cell growth and disease burden in patients. Since CD138 is cleaved by metalloproteinases and heparanase, soluble CD138 (sCD138) is detected in patient serum samples and its levels are associated with the prognosis of MM, with shorter survival in patients with higher levels [50]. Significantly, shedding of CD138 (sCD138) from MM cells stimulates myeloma cell growth by positive regulation and interaction with other MM-promoting factors (i.e., IL-6, vascular endothelial growth factor (VEGF), APRIL) in the BM microenvironment [40][48][51].
2.4. B-Cell Maturation Antigen (BCMA)
BCMA, also called tumor necrosis factor receptor superfamily member 17 (TNFRS17) or CD269, is a type III transmembrane protein with extracellular domains rich in cysteine without a signal peptide. BCMA, closely related to B-cell activation factor receptor (BAFF-R), and transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), regulates B cell proliferation and survival, as well as maturation and differentiation into plasma cells [52][53][54]. These three functionally related receptors bind to their cognate ligands, BAFF and/or APRIL, with different affinities, to support long-term survival of B cells at different stages of development. Specifically, BCMA, but not BAFF-R or TACI, is crucial for the long-term survival of plasma cells, but not overall B cell homeostasis [54]. During the differentiation of B cells into plasma cells, the expression of BCMA is induced from late memory cell, while BAFF-R is concomitantly extinguished. BCMA expression is regulated by B-lymphocyte-induced maturation protein 1 (BLIMP1), an important plasma cell transcriptional factor [55]. Under normal physiological conditions, the membrane BCMA is cleaved by gamma-secretase to form the soluble BCMA (sBCMA) [56]. Serum levels of sBCMA are significantly higher in MM patients than healthy individuals and associated with immune deficiency in the tumor microenvironment [57][58]. Elevated sBCMA levels are positively linked to increased tumor burden and poorer overall or progression-free survival [58]. Furthermore, the post-treatment levels of sBCMA could be used as a predictive marker for treatment response [59][60][61].
3. Other MM Tumor Antigens for Emerging Targeted Immunotherapy
The success of recent CAR-T, BiTE, and ADC, based on BCMA-targeting therapies, has quickly stimulated further development of immunotherapy targeting other novel antigens. Data of clinical studies further confirm that exclusive and high expression of tumor antigens on cancer cell is a key factor for new target selection to maximize the potency while minimizing the risk of off-target toxicity. Orphan G protein-coupled receptor, class C group 5 member D (GPRC5D), is a newly identified MM antigen that is highly expressed on MM cells in the BM but not normal tissue, although weakly expressed in hair follicles [62]. GPRC5D CAR-T exhibiting potent anti-MM activity in a preclinical study has led to ongoing clinical studies. BiAbs targeting GPRC5D and CD3 (talquetamab/JNJ-64407564 and GPRC5D TRAB) have also shown potent T-cell-mediated killing of GPRC5D+ MM cells and proliferation/activation of T cells in the preclinical and ongoing clinical studies [63][64]. Furthermore, the expression level of GPRC5D on MM cells and the BM microenvironment-related factors contribute to a different degree of responses to JNJ-7564 [65]. The early phase clinical trials of talquetamab (JNJ-64407564), as a monotherapy (NCT03399799) or combined with other anti-MM agents (NCT04108195), are ongoing, with already significant clinical activity.
Another potential antigen, integrin β7 (ITGB7), is associated with adhesion of MM cells to extra-cellular matrix elements, migration, invasion, and drug resistance [66]. In the in vitro study, novel ITGB7 targeting MMG49-derived CAR T cells showed specific MM cell lysis without damaging normal hematopoietic cells [67].
Natural Killer Group 2D (NKG2D) ligand, expressed on about 80% of MM cells, can bind to NKG2D on natural killer cells, leading to immune escape and tumor growth [68][69]. A BiAb targeting NKG2D and CS1 showed significant immune synapse between CS1+ MM cells and NKG2D+ immune cells, leading to effective MM lysis [70]. NKG2D-CAR T cells was also evaluated in a clinical trial, which showed good safety, but no objective response was observed (NCT02203825) [71].
References
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Spicka, I.; Oriol, A.; Hajek, R.; Rosinol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152.
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634.
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): A randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 781–794.
- Al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.L.; Mohty, M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2019, 9, 44.
- Nishimura, K.K.; Barlogie, B.; van Rhee, F.; Zangari, M.; Walker, B.A.; Rosenthal, A.; Schinke, C.; Thanendrarajan, S.; Davies, F.E.; Hoering, A.; et al. Long-term outcomes after autologous stem cell transplantation for multiple myeloma. Blood Adv. 2020, 4, 422–431.
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia 2017, 31, 1915–1921.
- Kumar, S.K.; Dimopoulos, M.A.; Kastritis, E.; Terpos, E.; Nahi, H.; Goldschmidt, H.; Hillengass, J.; Leleu, X.; Beksac, M.; Alsina, M.; et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: A multicenter IMWG study. Leukemia 2017, 31, 2443–2448.
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in biology of multiple myeloma: Clinical applications. Blood 2004, 104, 607–618.
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603.
- Tai, Y.T.; Cho, S.F.; Anderson, K.C. Osteoclast Immunosuppressive Effects in Multiple Myeloma: Role of Programmed Cell Death Ligand 1. Front. Immunol. 2018, 9, 1822.
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300.
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394.
- Zhang, L.; Tai, Y.T.; Ho, M.; Xing, L.; Chauhan, D.; Gang, A.; Qiu, L.; Anderson, K.C. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. 2017, 7, e547.
- Gorgun, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987.
- De Beule, N.; de Veirman, K.; Maes, K.; de Bruyne, E.; Menu, E.; Breckpot, K.; de Raeve, H.; van Rampelbergh, R.; van Ginderachter, J.A.; Schots, R.; et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J. Pathol. 2017, 241, 534–546.
- Chauhan, D.; Singh, A.V.; Brahmandam, M.; Carrasco, R.; Bandi, M.; Hideshima, T.; Bianchi, G.; Podar, K.; Tai, Y.T.; Mitsiades, C.; et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: A therapeutic target. Cancer Cell 2009, 16, 309–323.
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598.
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236.
- Tai, Y.T.; Anderson, K.C. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin. Biol. Ther. 2019, 19, 1143–1156.
- Neri, P.; Bahlis, N.J.; Lonial, S. New Strategies in Multiple Myeloma: Immunotherapy as a Novel Approach to Treat Patients with Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5959–5965.
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631.
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766.
- Pazina, T.; James, A.M.; MacFarlane, A.W., IV; Bezman, N.A.; Henning, K.A.; Bee, C.; Graziano, R.F.; Robbins, M.D.; Cohen, A.D.; Campbell, K.S. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology 2017, 6, e1339853.
- Tai, Y.T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337.
- Awwad, M.H.S.; Mahmoud, A.; Bruns, H.; Echchannaoui, H.; Kriegsmann, K.; Lutz, R.; Raab, M.S.; Bertsch, U.; Munder, M.; Jauch, A.; et al. Selective elimination of immunosuppressive T cells in patients with multiple myeloma. Leukemia 2021, 35, 2602–2615.
- Frasca, L.; Fedele, G.; Deaglio, S.; Capuano, C.; Palazzo, R.; Vaisitti, T.; Malavasi, F.; Ausiello, C.M. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood 2006, 107, 2392–2399.
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402.
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059.
- Lee, H.C.; Aarhus, R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157.
- Lee, H.C.; Aarhus, R.; Graeff, R.M. Sensitization of calcium-induced calcium release by cyclic ADP-ribose and calmodulin. J. Biol. Chem. 1995, 270, 9060–9066.
- Kim, S.Y.; Cho, B.H.; Kim, U.H. CD38-mediated Ca2+ signaling contributes to angiotensin II-induced activation of hepatic stellate cells: Attenuation of hepatic fibrosis by CD38 ablation. J. Biol. Chem. 2010, 285, 576–582.
- Kennedy, B.E.; Sadek, M.; Elnenaei, M.O.; Reiman, A.; Gujar, S.A. Targeting NAD(+) Synthesis to Potentiate CD38-Based Immunotherapy of Multiple Myeloma. Trends Cancer 2020, 6, 9–12.
- Mediero, A.; Cronstein, B.N. Adenosine and bone metabolism. Trends Endocrinol. Metab. 2013, 24, 290–300.
- Buckley, K.A.; Hipskind, R.A.; Gartland, A.; Bowler, W.B.; Gallagher, J.A. Adenosine triphosphate stimulates human osteoclast activity via upregulation of osteoblast-expressed receptor activator of nuclear factor-kappa B ligand. Bone 2002, 31, 582–590.
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Decombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derre, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J. Immunother. Cancer 2019, 7, 257.
- Boles, K.S.; Mathew, P.A. Molecular cloning of CS1, a novel human natural killer cell receptor belonging to the CD2 subset of the immunoglobulin superfamily. Immunogenetics 2001, 52, 302–307.
- Bouchon, A.; Cella, M.; Grierson, H.L.; Cohen, J.I.; Colonna, M. Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J. Immunol. 2001, 167, 5517–5521.
- Kumaresan, P.R.; Lai, W.C.; Chuang, S.S.; Bennett, M.; Mathew, P.A. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol. Immunol. 2002, 39, 1–8.
- Lee, J.K.; Mathew, S.O.; Vaidya, S.V.; Kumaresan, P.R.; Mathew, P.A. CS1 (CRACC, CD319) induces proliferation and autocrine cytokine expression on human B lymphocytes. J. Immunol. 2007, 179, 4672–4678.
- Wijdenes, J.; Vooijs, W.C.; Clement, C.; Post, J.; Morard, F.; Vita, N.; Laurent, P.; Sun, R.X.; Klein, B.; Dore, J.M. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br. J. Haematol. 1996, 94, 318–323.
- Shi, S.; Zhong, D.; Xiao, Y.; Wang, B.; Wang, W.; Zhang, F.; Huang, H. Syndecan-1 knockdown inhibits glioma cell proliferation and invasion by deregulating a c-src/FAK-associated signaling pathway. Oncotarget 2017, 8, 40922–40934.
- Kwon, M.J.; Jang, B.; Yi, J.Y.; Han, I.O.; Oh, E.S. Syndecans play dual roles as cell adhesion receptors and docking receptors. FEBS Lett. 2012, 586, 2207–2211.
- Altemeier, W.A.; Schlesinger, S.Y.; Buell, C.A.; Parks, W.C.; Chen, P. Syndecan-1 controls cell migration by activating Rap1 to regulate focal adhesion disassembly. J. Cell Sci. 2012, 125, 5188–5195.
- Chen, K.; Williams, K.J. Molecular mediators for raft-dependent endocytosis of syndecan-1, a highly conserved, multifunctional receptor. J. Biol. Chem. 2013, 288, 13988–13999.
- Yao, W.; Rose, J.L.; Wang, W.; Seth, S.; Jiang, H.; Taguchi, A.; Liu, J.; Yan, L.; Kapoor, A.; Hou, P.; et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 2019, 568, 410–414.
- Saleh, M.E.; Gadalla, R.; Hassan, H.; Afifi, A.; Gotte, M.; El-Shinawi, M.; Mohamed, M.M.; Ibrahim, S.A. The immunomodulatory role of tumor Syndecan-1 (CD138) on ex vivo tumor microenvironmental CD4+ T cell polarization in inflammatory and non-inflammatory breast cancer patients. PLoS ONE 2019, 14, e0217550.
- Szatmari, T.; Dobra, K. The role of syndecan-1 in cellular signaling and its effects on heparan sulfate biosynthesis in mesenchymal tumors. Front. Oncol. 2013, 3, 310.
- Akhmetzyanova, I.; McCarron, M.J.; Parekh, S.; Chesi, M.; Bergsagel, P.L.; Fooksman, D.R. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020, 34, 245–256.
- Andersen, N.F.; Standal, T.; Nielsen, J.L.; Heickendorff, L.; Borset, M.; Sorensen, F.B.; Abildgaard, N. Syndecan-1 and angiogenic cytokines in multiple myeloma: Correlation with bone marrow angiogenesis and survival. Br. J. Haematol. 2005, 128, 210–217.
- Seidel, C.; Sundan, A.; Hjorth, M.; Turesson, I.; Dahl, I.M.; Abildgaard, N.; Waage, A.; Borset, M. Serum syndecan-1: A new independent prognostic marker in multiple myeloma. Blood 2000, 95, 388–392.
- Dhodapkar, M.V.; Kelly, T.; Theus, A.; Athota, A.B.; Barlogie, B.; Sanderson, R.D. Elevated levels of shed syndecan-1 correlate with tumour mass and decreased matrix metalloproteinase-9 activity in the serum of patients with multiple myeloma. Br. J. Haematol. 1997, 99, 368–371.
- Tai, Y.T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199.
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821.
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98.
- Deng, S.; Yuan, T.; Cheng, X.; Jian, R.; Jiang, J. B-lymphocyte-induced maturation protein1 up-regulates the expression of B-cell maturation antigen in mouse plasma cells. Mol. Biol. Rep. 2010, 37, 3747–3755.
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rubsamen, H.; et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333.
- Sanchez, E.; Gillespie, A.; Tang, G.; Ferros, M.; Harutyunyan, N.M.; Vardanyan, S.; Gottlieb, J.; Li, M.; Wang, C.S.; Chen, H.; et al. Soluble B-Cell Maturation Antigen Mediates Tumor-Induced Immune Deficiency in Multiple Myeloma. Clin. Cancer Res. 2016, 22, 3383–3397.
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795.
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Invest. 2019, 129, 2210–2221.
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280.
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kronke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783.
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11.
- Pillarisetti, K.; Edavettal, S.; Mendonca, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243.
- Kodama, T.; Kochi, Y.; Nakai, W.; Mizuno, H.; Baba, T.; Habu, K.; Sawada, N.; Tsunoda, H.; Shima, T.; Miyawaki, K.; et al. Anti-GPRC5D/CD3 Bispecific T-Cell-Redirecting Antibody for the Treatment of Multiple Myeloma. Mol. Cancer Ther. 2019, 18, 1555–1564.
- Verkleij, C.P.M.; Broekmans, M.E.C.; van Duin, M.; Frerichs, K.A.; Kuiper, R.; de Jonge, A.V.; Kaiser, M.; Morgan, G.; Axel, A.; Boominathan, R.; et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv. 2021, 5, 2196–2215.
- Neri, P.; Ren, L.; Azab, A.K.; Brentnall, M.; Gratton, K.; Klimowicz, A.C.; Lin, C.; Duggan, P.; Tassone, P.; Mansoor, A.; et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 2011, 117, 6202–6213.
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S.; et al. The activated conformation of integrin beta7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat. Med. 2017, 23, 1436–1443.
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005, 105, 251–258.
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449.
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787.
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112.
More
Information
Subjects:
Oncology; Immunology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
862
Revisions:
2 times
(View History)
Update Date:
27 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No