Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yu-Tzu Tai and Version 2 by Yvaine Wei.
The approval of monoclonal antibody (MoAb) against CD38 (daratumumab) and SLAMF7 (elotuzumab) in relapsed and refractory MM (RRMM) represents an important milestone in the development of targeted immunotherapy in MM. These MoAb-based agents significantly induce cytotoxicity of MM cells via multiple effector-dependent mechanisms and can further induce immunomodulation to repair a dysfunctional tumor immune microenvironment.
- multiple myeloma
- MM
- immunotherapy
- immunomodulatory drugs
- IMiDs
- MoAb
- CD38
- SLAMF7
- BCMA
- bone marrow (BM) microenvironment
- GPRC5D
1. Introduction
The development and introduction of the proteasome inhibitor (PI) bortezomib and immunomodulatory drugs (IMiDs), including thalidomide and lenalidomide, has revolutionized the treatment paradigm for multiple myeloma (MM). Second-generation drugs within the same classes, such as carfilzomib and ixazomib (PIs) and pomalidomide (IMiDs), further improve the response rate, survival, and safety profile [1][2][3][1,2,3]. The incorporation of autologous stem cell transplantation in eligible patients has also prolonged survival with more durable disease control [4][5][4,5]. However, disease recurrence remains common for most MM patients. Since drug-resistant clones constantly emerge and evolve, leading to a low 5-year overall survival rate in real-world data [6]. The clinical outcomes of patients with relapsed or refractory MM (RRMM) are dismally poor because of the gradually decreased durability of the response to successive lines of anti-MM therapy [7].
Accumulating studies for the past decades have defined that the bone marrow (BM) microenvironment is essential in supporting MM cell growth, survival, and drug resistance. MM cells are in close contact with surrounding BM accessory cells through bi-directional interactions, including stromal cells (BMSCs) [8], osteoclasts (OCs) [9][10][9,10], regulatory T (Treg) or B (Breg) cells [11][12][13][11,12,13], myeloid-derived suppressor cells (MDSCs) [14], tumor-associated macrophages (TAMs) [15], and plasmacytoid dendritic cells [16]. These non-MM cells, in turn, secrete abnormal levels of a variety of cytokines and growth factors in a paracrine fashion to promote pathogenesis of MM, including interleukin-6 (IL-6), IL-10, MIP-1α/β, transforming growth factor-beta (TGFβ), stromal cell-derived factor-1 (SDF-1), and a proliferation-inducing ligand (APRIL) [9][17][18][19][9,17,18,19]. Furthermore, changes in BM accessory cells and cytokines, either secreted by accessory cells or MM cells via autocrine or paracrine manners, contribute to myeloma cell immune escape, inhibition of myeloma-specific T effector cells, induction of T-cell anergy, and abnormality in Treg cells, resulting in an immunosuppressive microenvironment that impairs immunotherapy [20].
Monoclonal antibodies (MoAbs) binding to selective molecules on the surface of cancer cells have transformed cancer treatment. In principle, these biologically based molecules/proteins induce tumor cell killing mainly dependent on effector functions, including antibody-dependent cellular cytotoxicity (ADCC) via CD16-expressing effector cells (i.e., NK cells, neutrophils, monocytes), complement-dependent cytotoxicity (CDC), and/or antibody-dependent cellular phagocytosis (ADCP) via macrophages. These primary mechanisms of action are distinct from small molecules used in conventional chemotherapies, which directly induce tumor cell apoptosis and are largely independent of immune effector function. The first two therapeutic MoAbs available for RRMM patients are MoAbs targeting CD38 (daratumumab) and SLAMF7 (also named CS1) (elotuzumab), approved by the U.S. Food and Drug Administration (FDA) in 2015 [21][22][21,22]. These represent an important breakthrough for effective targeted immune-based therapies in MM. Importantly, results obtained from preclinical and clinical studies of both MoAbs thus far have shown that these first-generation targeting bio-molecules also affect the immunosuppressive non-MM cell components in addition to MM cells [11][12][13][23][24][25][11,12,13,23,24,25]. These findings have inspired many investigations on identifying the patho-immunological roles of various immune regulatory cell subsets and molecules regulating their function using in vitro, ex vivo, and in vivo models.
2. Pathophysiological Function for Validated MM Target Antigens and Their Related Immunotherapies
The anti-MM mechanisms of targeted immunotherapeutic bio-reagents are mainly derived from the MoAbs, which detect and engage with selective proteins on MM cell membrane followed by the induction of NK cell-mediated killing (Figure 1 and Figure 2). These target antigens are chosen based on their differential expression and/or critical roles in growth, survival, and drug resistance in MM cells.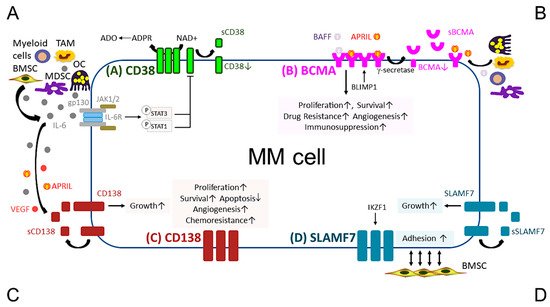
Figure 1. Pathobiological function of validated MM target antigens commonly used in current MM immunotherapies. (A) CD38, a receptor and an ectoenzyme, regulates the conversion of NAD+ to ADO, which contributes to the immunocompromised tumor microenvironment. IL-6, an important MM growth, survival, and immunosuppressive cytokine, mainly produced by non-myeloma BM cells (i.e., BMSCs, MDSC, OC, and TAM), binds to IL-6R, which interacts with gp130 to activate the JAK1/2-STAT1/3 signaling pathway and subsequently downregulates CD38 expression in MM cells. (B) BCMA is the cognate receptor for APRIL and BAFF, which are present in the BM microenvironment. APRIL, an important plasma cell factor, preferentially binds to BCMA when compared with BAFF, to induce signaling pathways critical to promote survival, proliferation, and drug resistance, as well as immunosuppression of MM cells. BCMA expression is induced by BLIMP1, a key plasma cell transcriptional regulator, and BCMA protein is cleaved by gamma (γ)-secretase, resulting in the soluble form (sBCMA) that is detected in MM patient serum and correlated with disease advancement. (C) CD138, the transmembrane heparan sulfate proteoglycan syndecan-1, is overexpressed in malignant plasma cells and its levels are associated with increased proliferation and survival, as well as decreased apoptosis in MM cell lines and patient MM cells. Levels of shed CD138 (sCD138) cleaved by metalloproteinases and heparanase in MM patient serum samples are also linked to disease progression and sCD138 acts locally or distally with various effector molecules (i.e., IL-6, APRIL, and VEGF) to impact tumor progression. (D) SLAMF7, also named CS1, promotes adhesion of MM cells to BMSCs. The expression of SLAMF7 is regulated by the transcriptional factor IKZF1 (Ikaros), a MM-related target by lenalidomide and pomalidomide, in MM cells. Soluble SLAMF7 (sSLAMF7) is detected in patient serum and promotes MM cell growth via homophilic interaction with SLAMF7 on MM cells. Among these four MM antigens, BCMA has the most limited expression at the transcript and protein levels in plasma cells but no other normal tissues except a minute subset of plasmacytoid dendritic cell. ADO, adenosine; ADPR, ADP-ribose; APRIL, A proliferation inducing ligand; BAFF, B-cell activating factor; BMSC, bone marrow stromal cell; JAK, Janus kinase; MDSC, myeloid-derived suppressor cell; NAD+, nicotinamide adenine dinucleotide; OC, osteoclast; TAM, tumor-associated macrophages; VEGF, vascular endothelial growth factor.
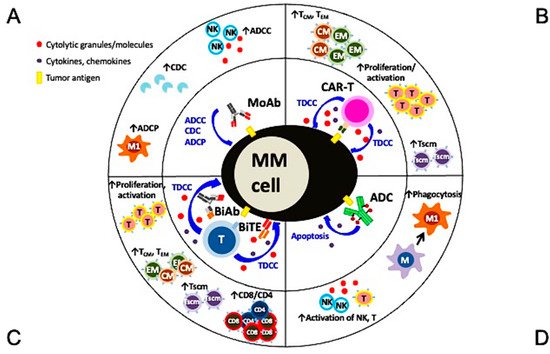
Figure 2. Multiple mechanisms of action and immunomodulatory effects of current targeted immunotherapies in MM. (A) Monoclonal antibodies (MoAbs) identify and bind to specific antigens to trigger MM cell lysis via multiple immune-dependent mechanisms, including ADCC, ADCP, and CDC. (B) The single chain fragment variant of the CAR-T cell first targets the MM-specific antigen and potent MM cell lysis is induced in a major histocompatibility complex-independent manner followed by increased proliferation and activation of T effector cells, including those with central and effector memory phenotypes. (C) BiAb or BiTE, off-shelf products different from autologous CAR-T cells, simultaneously binds to specific tumor antigen on MM cells and CD3 on T cells to trigger potent MM cell killing as well as proliferation of immune effector T cells. T cells with memory phenotypes (Tcm, Tem, Tscm) and the major cytolytic T cells (CD8+ cells) are increased significantly. (D) The MoAb portion of antibody drug conjugate (ADC) binds to tumor antigen on MM cell membrane and the ADC is endocytosed followed by the release of potent cytotoxic payloads from lysosomes to directly induce MM cell apoptosis. Payloads used in ADC are typically even more potent than those used in chemotherapies to improve superior specific killing of tumors cells but not the surrounding normal tissues. Depending on the design of the ADC, NK cells and macrophages are directly or indirectly activated to induce ADCC and phagocytosis, respectively, to further augment killing of tumor cells. ADCC, antibody-dependent cellular cytotoxicity (NK cells are the predominant effector cells. Other CD16-expressing effectors including monocytes and neutrophils are also capable to induce ADCC); ADCP, antibody-dependent cellular phagocytosis (macrophage (M), especially with the M1 characteristics, are the key effector cells whereas macrophages with M2 features (TAM in Figure 1) promote tumor growth); BiAb, bispecific antibody engaging with T cells; BiTE, bispecific T cell engager; CDC, complement-dependent cytotoxicity; Tcm, central memory T cell; Tem, effector memory T cell; Tscm, stem cell-like memory T cell.
2.1. CD38
CD38, a type II transmembrane glycoprotein, was first identified as a marker of cell activation and proliferation in lymphocytes. It regulates cell migration [26][29] and receptor-mediated adhesion via interaction with endothelial CD31 or hyaluronic acid [27][30]. CD38 receptor also exhibits ecto-enzymatic activity, involved in the metabolism of cytoplasmic nicotinamide adenine dinucleotide phosphate (NADP) and extracellular nicotinamide adenine dinucleotide (NAD+) [28][31]. In addition, CD38 interacts with its substrate, NAD+, to increase production of Ca2+ mobilizing compounds; i.e., cyclic adenosine diphosphate ribose (CADPR), ADP ribose (ADPR), and nicotinic acid adenine dinucleotide phosphate [29][30][31][32,33,34]. ADPR is further converted to adenosine monophosphate (AMP) by CD203a and then adenosine (ADO), which exhibits immunosuppressive activity via a reduction in immune cell activity and induction of differentiation of osteoclast, one of the most immunosuppressive BM accessory cells [32][33][34][35][35,36,37,38]. Moreover, the malignant plasma cells further use aerobic glycolysis to promote an acidic BM, which together with CD38 highly expressed on their surface induce the generation of AMP and ADO.