 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gianluigi Mazzoccoli | + 6231 word(s) | 6231 | 2021-12-23 07:46:13 | | | |
| 2 | Vivi Li | Meta information modification | 6231 | 2021-12-24 03:02:57 | | |
Video Upload Options
There is an under-recognized role of the aryl hydrocarbon receptor (AhR) in co-ordinating the entry and pathophysiology of the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) that underpins the COVID-19 pandemic. The rise in pro-inflammatory cytokines during the ‘cytokine storm’ induce indoleamine 2,3-dioxygenase (IDO), leading to an increase in kynurenine that activates the AhR, thereby heightening the initial pro-inflammatory cytokine phase and suppressing the endogenous anti-viral response. Such AhR-driven changes underpin the heightened severity and fatality associated with pre-existent high-risk medical conditions, such as type II diabetes, as well as to how racial discrimination stress contributes to the raised severity/fatality in people from the Black Asian and Minority Ethnic (BAME) communities. The AhR is pivotal in modulating mitochondrial metabolism and co-ordinating specialized, pro-resolving mediators (SPMs), the melatonergic pathways, acetyl-coenzyme A, and the cyclooxygenase (COX) 2-prostaglandin (PG) E2 pathway that underpin ‘exhaustion’ in the endogenous anti-viral cells, paralleling similar metabolic suppression in cytolytic immune cells that is evident across all cancers.
1. Introduction
The Aryl Hydrocarbon Receptor
2. SARS-CoV-2 Entry and Pathophysiology
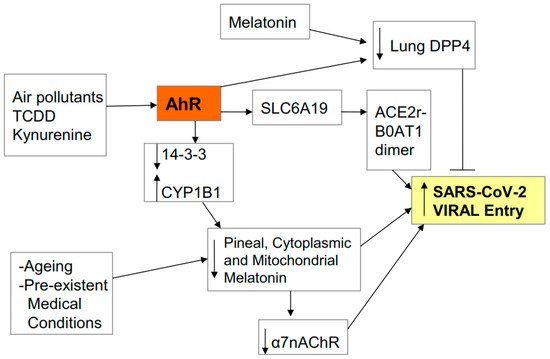
2.1. Entry
2.2. Pathophysiology
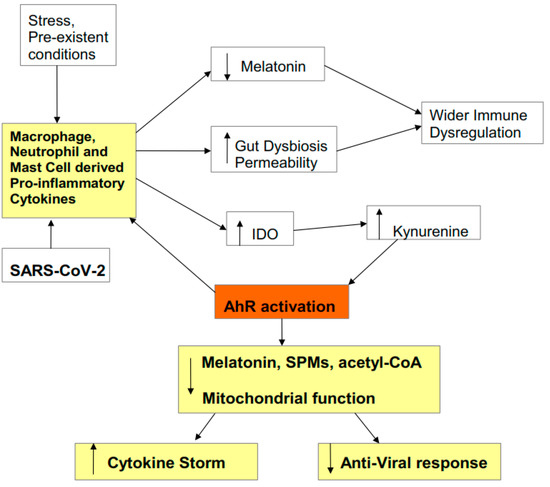
2.3. Cytokine Storm Consequences
2.4. AhR Regulation of Mitochondrial Metabolism
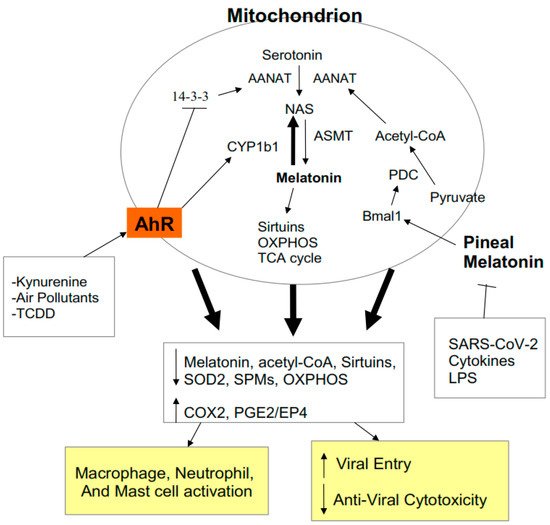
2.5. AhR and Pre-Existing High-Risk COVID-19 Medical Conditions
2.6. AhR and Stress
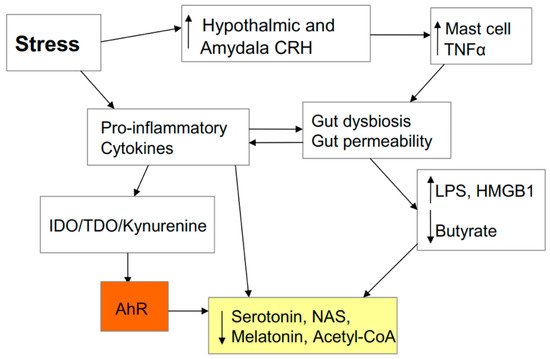
2.7. Stress and the Gut
3. AhR and Wider COVID-19 Pathophysiology
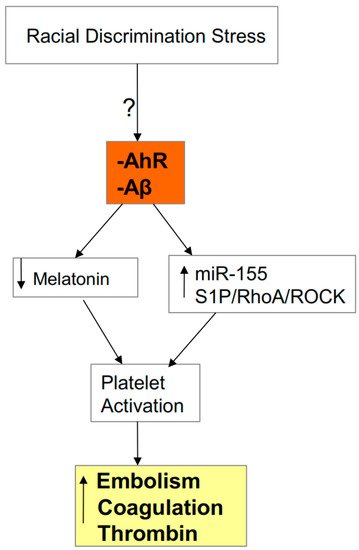
3.1. AhR, Platelets, ROCK, and SARS-CoV-2 Severity/Fatality
3.2. AhR, Acetyl-CoA, COX2, and Specialized Pro-Resolving Mediators (SPMs)

3.3. AhR, COX2, SPMs, Acetyl-CoA, and miR-155
3.4. Gut Dysbiosis: Interactions with Acetyl-CoA, COX2, SPMs, and AhR
References
- Kloc, M.; Ghobrial, R.M.; Kubiak, J.Z. How nicotine can inhibit cytokine storm in the lungs and prevent or lessen the severity of COVID-19 infection? Immunol. Lett. 2020, 224, 28–29.
- Potter, H.; Boyd, T.D.; Clarke, P.; Pelak, V.S.; Tyler, K.L. Recruiting the innate immune system with GM-CSF to fight viral diseases, including West Nile Virus encephalitis and COVID-19. F1000Research 2020, 9, 345.
- Chakrabarti, S. Focusing On a Unique Innate Memory Cell Population of Natural Killer Cells in the Fight Against COVID-19: Harnessing The Ubiquity Of Cytomegalovirus Exposure. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020047.
- Fierabracci, A.; Arena, A.; Rossi, P. COVID-19: A Review on Diagnosis, Treatment, and Prophylaxis. Int. J. Mol. Sci. 2020, 21, 5145.
- Anderson, G.; Reiter, R.J. COVID-19 pathophysiology: Interactions of gut microbiome, melatonin, vitamin D, stress, kynurenine and the alpha 7 nicotinic receptor: Treatment implications. Melatonin Res. 2020, 3, 322–345.
- Küster, O.C.; Laptinskaya, D.; Fissler, P.; Schnack, C.; Zügel, M.; Nold, V.; Thurm, F.; Pleiner, S.; Karabatsiakis, A.; Von Einem, B.; et al. Novel Blood-Based Biomarkers of Cognition, Stress, and Physical or Cognitive Training in Older Adults at Risk of Dementia: Preliminary Evidence for a Role of BDNF, Irisin, and the Kynurenine Pathway. J. Alzheimer’s Dis. 2017, 59, 1097–1111.
- Nduhirabandi, F.; Du Toit, E.F.; Lochner, A. Melatonin and the metabolic syndrome: A tool for effective therapy in obesity-associated abnormalities? Acta Physiol. 2012, 205, 209–223.
- Zhao, Y.; Fu, Y.; Sun, Y.; Zou, M.; Peng, X. Transcriptional Regulation of gga-miR-451 by AhR: Arnt in Mycoplasma gallisepticum (HS Strain) Infection. Int. J. Mol. Sci. 2019, 20, 3087.
- Raisi-Estabragh, Z.; McCracken, C.; Bethell, M.S.; Cooper, J.; Cooper, C.; Caulfield, M.J.; Munroe, P.B.; Harvey, N.C.; Petersen, S.E. Greater risk of severe COVID-19 in Black, Asian and Minority Ethnic populations is not explained by cardiometabolic, socioeconomic or behavioural factors, or by 25(OH)-vitamin D status: Study of 1326 cases from the UK Biobank. J. Public Health 2020, 42.
- Yan, C.; Luo, Z.; Li, W.; Li, X.; Dallmann, R.; Kurihara, H.; Li, Y.; He, R.-R. Disturbed Yin–Yang balance: Stress increases the susceptibility to primary and recurrent infections of herpes simplex virus type 1. Acta Pharm. Sin. B 2020, 10, 383–398.
- Cohen, S.; Tyrrell, D.A.; Smith, A.P. Psychological Stress and Susceptibility to the Common Cold. N. Eng. J. Med. 1991, 325, 606–612.
- Scalise, M.; Indiveri, C. Repurposing Nimesulide, a Potent Inhibitor of the B0AT1 Subunit of the SARS-CoV-2 Receptor, as a Therapeutic Adjuvant of COVID-19. SLAS Discov. Adv. Sci. Drug Discov. 2020, 2472555220934421.
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448.
- Javed, K.; Cheng, Q.; Carroll, A.J.; Truong, T.T.; Bröer, S. Development of Biomarkers for Inhibition of SLC6A19 (B0AT1)—A Potential Target to Treat Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 3597.
- Tian, W.; Fu, H.; Xu, T.; Xu, S.L.; Guo, Z.; Tian, J.; Tao, W.; Xie, H.Q.; Zhao, B. SLC6A19 is a novel putative gene, induced by dioxins via AhR in human hepatoma HepG2 cells. Environ. Pollut. 2018, 237, 508–514.
- Wang, Y.; Han, D.; Zhou, T.; Zhang, J.; Liu, C.; Cao, F.; Dong, N. Melatonin ameliorates aortic valve calcification via the regulation of circular RNA CircRIC3/miR-204-5p/DPP4 signaling in valvular interstitial cells. J. Pineal Res. 2020, 69, 12666.
- Lv, D.; Xu, Y.; Cheng, H.; Ke, Y.; Zhang, X.; Ying, K. A novel cell-based assay for dynamically detecting neutrophil extracellular traps-induced lung epithelial injuries. Exp. Cell Res. 2020, 394, 112101.
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rorsales-Corral, S.; Chuffa, L.G.D.A. Melatonin inhibits Warburg-dependent cancer by redirecting glucose oxidation to the mitochondria: A mechanistic hypothesis. Cell. Mol. Life Sci. 2020, 77, 2527–2542.
- Anderson, G.; Rodriguez, M.; Reiter, R.J. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. Int. J. Mol. Sci. 2019, 20, 5500.
- Guo, M.; Wang, H.; Xu, S.; Zhuang, Y.; An, J.; Su, C.; Xia, Y.; Chen, J.; Xu, Z.Z.; Liu, Q.; et al. Alteration in gut microbiota is associated with dysregulation of cytokines and glucocorticoid therapy in systemic lupus erythematosus. Gut Microbes 2020, 11, 1–16.
- Zuo, T.; Zhang, F.; Lui, G.C.; Yeoh, Y.K.; Li, A.Y.; Zhan, H.; Wan, Y.; Chung, A.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020.
- Vitale, J.A.; Perazzo, P.; Silingardi, M.; Biffi, M.; Banfi, G.; Negrini, F. Is disruption of sleep quality a consequence of severe Covid-19 infection? A case-series examination. Chronobiol. Int. 2020.
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, 140327.
- Anderson, G.; Maes, M. Mitochondria and immunity in chronic fatigue syndrome. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2020, 103, 109976.
- Anderson, G.; Mazzoccoli, G. Left Ventricular Hypertrophy: Roles of Mitochondria CYP1B1 and Melatonergic Pathways in Co-Ordinating Wider Pathophysiology. Int. J. Mol. Sci. 2019, 20, 4068.
- Muxel, S.M.; Lapa, M.; Monteiro, A.W.A.; Cecon, E.; Tamura, E.K.P.; Flöeter-Winter, L.M.; Markus, R.P. NF-κB Drives the Synthesis of Melatonin in RAW 264.7 Macrophages by Inducing the Transcription of the Arylalkylamine-N-Acetyltransferase (AA-NAT) Gene. PLoS ONE 2012, 7, e52010.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 10.
- Pfeifer, C.; Highton, A.J.; Peine, S.; Sauter, J.; Schmidt, A.H.; Bunders, M.J.; Altfeld, M.; Körner, C. Natural Killer Cell Education Is Associated With a Distinct Glycolytic Profile. Front. Immunol. 2018, 9, 3020.
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Boil. 2020, 108, 17–41.
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Acuna-Castroviejo, D.; Escames, G. Inhibition of mitochondrial pyruvate dehydrogenase kinase: A proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res. 2019, 2, 105–119.
- Anderson, G. Daytime orexin and night-time melatonin regulation of mitochondria melatonin: Roles in circadian oscillations systemically and centrally in breast cancer symptomatology. Melatonin Res. 2019, 2, 1–8.
- Acuña-Castroviejo, D.; Rahim, I.; Acuña-Fernández, C.; Ortiz, F.; Solera-Marín, J.; Sayed, R.K.A.; Díaz-Casado, M.E.; Rusanova, I.; López, L.C.; Escames, G. Melatonin, clock genes and mitochondria in sepsis. Cell. Mol. Life Sci. 2017, 74, 3965–3987.
- Ren, S.; Correia, M.A. Heme: A Regulator of Rat Hepatic Tryptophan 2,3-Dioxygenase? Arch. Biochem. Biophys. 2000, 377, 195–203.
- Michels, N.; Clarke, G.; Olavarría-Ramírez, L.; Gomez-Martinez, S.; Díaz-Prieto, L.; Marcos, A.; Widhalm, K.; Carvalho, L.A. Psychosocial stress and inflammation driving tryptophan breakdown in children and adolescents: A cross-sectional analysis of two cohorts. Psychoneuroendocrinology 2018, 94, 104–111.
- Chiappelli, J.; Rowland, L.M.; Notarangelo, F.M.; Wijtenburg, S.A.; Thomas, M.A.R.; Pocivavsek, A.; Jones, A.; Wisner, K.; Kochunov, P.; Schwarcz, R.; et al. Salivary kynurenic acid response to psychological stress: Inverse relationship to cortical glutamate in schizophrenia. Neuropsychopharmacology 2018, 43, 1706–1711.
- Bettison, T.M.; Nahm, C.B.; Gill, A.J.; Mittal, A.; Malhi, G.S.; Samra, J.S. Understanding the Pathophysiology of Psychological Distress and Pancreatic Cancer. Pancreas 2018, 47, 376–381.
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites with Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan Res. 2017, 10.
- Sakurai, M.; Yamamoto, Y.; Kanayama, N.; Hasegawa, M.; Mouri, A.; Takemura, M.; Matsunami, H.; Miyauchi, T.; Tokura, T.; Kimura, H.; et al. Serum Metabolic Profiles of the Tryptophan-Kynurenine Pathway in the high risk subjects of major depressive disorder. Sci. Rep. 2020, 10, 1–13.
- Anderson, G. Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2018, 80, 255–266.
- Evrensel, A.; Ünsalver, B.Ö.; Ceylan, M.E. Immune-Kynurenine Pathways and the Gut Microbiota-Brain Axis in Anxiety Disorders. Adv. Exp. Med. Biol. 2020, 1191, 155–167.
- Petrozzi, B.P.; ElYamany, O.; Rummel, C.; Mulert, C. Effects of inflammation on the kynurenine pathway in schizophrenia—A systematic review. J. Neuroinflamm. 2020, 17, 1–17.
- Anderson, G. Neuronal–immune interactions in mediating stress effects in the etiology and course of schizophrenia: Role of the amygdala in developmental co-ordination. Med. Hypotheses 2011, 76, 54–60.
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top. Med. Chem. 2020, 20, 524–539.
- Vanuytsel, T.; Van Wanrooy, S.; Vanheel, H.; Vanormelingen, C.; Verschueren, S.; Houben, E.; Rasoel, S.S.; Toth, J.; Holvoet, L.; Farré, R.; et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut 2014, 63, 1293–1299.
- Martín-Hernández, D.; Tendilla-Beltran, H.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C.; Caso, J.R. Chronic Mild Stress Alters Kynurenine Pathways Changing the Glutamate Neurotransmission in Frontal Cortex of Rats. Mol. Neurobiol. 2019, 56, 490–501.
- Fuertig, R.; Azzinnari, D.; Bergamini, G.; Cathomas, F.; Sigrist, H.; Seifritz, E.; Vavassori, S.; Luippold, A.; Hengerer, B.; Ceci, A.; et al. Mouse chronic social stress increases blood and brain kynurenine pathway activity and fear behaviour: Both effects are reversed by inhibition of indoleamine 2,3-dioxygenase. Brain Behav. Immun. 2016, 54, 59–72.
- Nold, V.; Sweatman, C.; Karabatsiakis, A.; Böck, C.; Bretschneider, T.; Lawless, N.; Fundel-Clemens, K.; Kolassa, I.-T.; Allers, K.; Catherine, S. Activation of the kynurenine pathway and mitochondrial respiration to face allostatic load in a double-hit model of stress. Psychoneuroendocrinology 2019, 107, 148–159.
- Wang, B.; Lian, Y.-J.; Su, W.-J.; Peng, W.; Dong, X.; Liu, L.-L.; Gong, H.-Y.; Zhang, T.; Jiang, C.-L.; Wang, Y.-X. HMGB1 mediates depressive behavior induced by chronic stress through activating the kynurenine pathway. Brain Behav. Immun. 2018, 72, 51–60.
- Marin, I.A.; Goertz, J.; Ren, T.; Rich, S.S.; Onengut-Gumuscu, S.; Farber, E.; Wu, M.; Overall, C.C.; Kipnis, J.; Gaultier, A. Microbiota alteration is associated with the development of stress-induced despair behavior. Sci. Rep. 2017, 7, 43859.
- Bompard, F.; Monnier, H.; Saab, I.; Tordjman, M.; Abdoul, H.; Fournier, L.; Sanchez, O.; Lorut, C.; Chassagnon, G.; Revel, M.-P. Pulmonary embolism in patients with COVID-19 pneumonia. Eur. Respir. J. 2020, 56, 2001365.
- Pombo, M.; Lame, M.W.; Walker, N.J.; Huynh, D.H.; Tablin, F. TCDD and omeprazole prime platelets through the aryl hydrocarbon receptor (AhR) non-genomic pathway. Toxicol. Lett. 2015, 235, 28–36.
- Wang, Y.-C.; Tsai, C.-F.; Chuang, H.-L.; Chang, Y.-C.; Chen, H.-S.; Lee, J.-N.; Tsai, E.-M. Benzyl butyl phthalate promotes breast cancer stem cell expansion via SPHK1/S1P/S1PR3 signaling. Oncotarget 2016, 7, 29563–29576.
- Wang, H.-C.; Wong, T.-H.; Wang, L.-T.; Su, H.-H.; Yu, H.-Y.; Wu, A.-H.; Lin, Y.-C.; Chen, H.-L.; Suen, J.-L.; Hsu, S.-H.; et al. Aryl hydrocarbon receptor signaling promotes ORMDL3-dependent generation of sphingosine-1-phosphate by inhibiting sphingosine-1-phosphate lyase. Cell. Mol. Immunol. 2019, 16, 783–790.
- Punsawad, C.; Viriyavejakul, P. Expression of sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 in malaria-associated acute lung injury/acute respiratory distress syndrome in a mouse model. PLoS ONE 2019, 14, e0222098.
- Xu, X.; Shi, L.; Ma, X.; Su, H.; Ma, G.; Wu, X.; Ying, K.; Zhang, R. RhoA-Rho associated kinase signaling leads to renin-angiotensin system imbalance and angiotensin converting enzyme 2 has a protective role in acute pulmonary embolism. Thromb. Res. 2019, 176, 85–94.
- Lu, Y.; Lian, Z.; Yang, H.; Jiang, Q.; Zou, Y.; Zhu, Y.; Ling, W.; Yuan, L.; Jiang, X.; Chen, S. TNF-α activates RhoA/ROCK signaling pathway and increases permeability of endothelial cells infected with Listeria monocytogenes. Chin. J. Cell. Infect. Mol. Immunol. 2020, 36, 193–197.
- Sonkar, V.K.; Kulkarni, P.; Dash, D. Amyloid β peptide stimulates platelet activation through RhoA-dependent modulation of actomyosin organization. FASEB J. 2014, 28, 1819–1829.
- Lee, J.Y.; Han, S.H.; Park, M.H.; Song, I.-S.; Choi, M.-K.; Yu, E.; Park, C.-M.; Kim, H.J.; Kim, S.H.; Schuchman, E.H.; et al. N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–19.
- Zhu, P.; Yu, H.; Zhou, K.; Bai, Y.; Qi, R.-Q.; Zhang, S.-G. 3,3′-Diindolylmethane modulates aryl hydrocarbon receptor of esophageal squamous cell carcinoma to reverse epithelial-mesenchymal transition through repressing RhoA/ROCK1-mediated COX2/PGE2 pathway. J. Exp. Clin. Cancer Res. 2020, 39, 1–18.
- Cheng, C.-I.; Chen, P.-H.; Lin, Y.-C.; Kao, Y.-H. High glucose activates Raw264.7 macrophages through RhoA kinase-mediated signaling pathway. Cell. Signal. 2015, 27, 283–292.
- Gdula-Argasińska, J.; Czepiel, J.; Totoń-Żurańska, J.; Jurczyszyn, A.; Wołkow, P.P.; Librowski, T.; Perucki, W. Resolvin D1 down-regulates CYP1A1 and PTGS2 gene in the HUVEC cells treated with benzo(a)pyrene. Pharmacol. Rep. 2016, 68, 939–944.
- Vogel, C.F.A.; Kado, S.Y.; Kobayashi, R.; Liu, X.; Wong, P.; Na, K.; Durbin, T.; Okamoto, R.A.; Kado, N.Y. Inflammatory marker and aryl hydrocarbon receptor-dependent responses in human macrophages exposed to emissions from biodiesel fuels. Chemosphere 2019, 220, 993–1002.
- Castañeda, A.R.; Pinkerton, K.E.; Bein, K.J.; Magaña-Méndez, A.; Yang, H.T.; Ashwood, P.; Vogel, C.F. Ambient particulate matter activates the aryl hydrocarbon receptor in dendritic cells and enhances Th17 polarization. Toxicol. Lett. 2018, 292, 85–96.
- Zavan, B.; De Almeida, E.M.; Salles, É.D.S.L.; Amarante-Paffaro, A.M.D.; Paffaro, V.A. COX-2 plays a role in angiogenic DBA + uNK cell subsets activation and pregnancy protection in LPS-exposed mice. Placenta 2016, 44, 34–45.
- Mitchell, J.A.; Shala, F.; Elghazouli, Y.; Warner, T.D.; Gaston-Massuet, C.; Crescente, M.; Armstrong, P.C.; Herschman, H.R.; Kirkby, N.S. Cell-Specific Gene Deletion Reveals the Antithrombotic Function of COX1 and Explains the Vascular COX1/Prostacyclin Paradox. Circ. Res. 2019, 125, 847–854.
- Ma, X.; Holt, D.; Kundu, N.; Reader, J.; Goloubeva, O.; Take, Y.; Fulton, A.M. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. OncoImmunology 2013, 2, e22647.
- Holt, D.; Ma, X.; Kundu, N.; Fulton, A.M. Prostaglandin E2 (PGE2) suppresses natural killer cell function primarily through the PGE2 receptor EP4. Cancer Immunol. Immunother. 2011, 60, 1577–1586.
- Eberstål, S.; Fritzell, S.; Sandén, E.; Visse, E.; Darabi, A.; Siesjö, P. Immunizations with unmodified tumor cells and simultaneous COX-2 inhibition eradicate malignant rat brain tumors and induce a long-lasting CD8+ T cell memory. J. Neuroimmunol. 2014, 274, 161–167.
- Tawfik, D.; Groth, C.; Gundlach, J.-P.; Peipp, M.; Kabelitz, D.; Becker, T.; Oberg, H.-H.; Trauzold, A.; Wesch, D. TRAIL-Receptor 4 Modulates γδ T Cell-Cytotoxicity Toward Cancer Cells. Front. Immunol. 2019, 10, 10.
- Inada, T.; Kubo, K.; Shingu, K. Promotion of interferon-gamma production by natural killer cells via suppression of murine peritoneal macrophage prostaglandin E2 production using intravenous anesthetic propofol. Int. Immunopharmacol. 2010, 10, 1200–1208.
- Haworth, O.; Cernadas, M.; Levy, B.D. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J. Immunol. 2011, 186, 6129–6135.
- Chia, N.; Kumar, R.K.; Foster, P.S.; Herbert, C. Enhanced Pro-Inflammatory Response of Macrophages to Interleukin-33 in an Allergic Environment. Int. Arch. Allergy Immunol. 2018, 176, 74–82.
- Barden, A.; Shinde, S.; Tsai, I.-J.; Croft, K.D.; Beilin, L.; Puddey, I.; Mori, T. Effect of weight loss on neutrophil resolvins in the metabolic syndrome. Prostaglandins Leukot. Essent. Fat. Acids 2019, 148, 25–29.
- Freire, M.; Dalli, J.; Serhan, C.N.; Van Dyke, T.E.; Charles, S.N. Neutrophil Resolvin E1 Receptor Expression and Function in Type 2 Diabetes. J. Immunol. 2017, 198, 718–728.
- Kang, G.-J.; Lee, H.-J.; Kang, Y.P.; Kim, E.J.; Kim, H.J.; Byun, H.J.; Park, M.K.; Cho, H.; Kwon, S.W.; Lee, C.-H. High-mobility group box 1 suppresses resolvin D1-induced phagocytosis via induction of resolvin D1-inactivating enzyme, 15-hydroxyprostaglandin dehydrogenase. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 1981–1988.
- Chiurchiù, V.; Leuti, A.; Saracini, S.; Fontana, D.; Finamore, P.; Giua, R.; Padovini, L.; Incalzi, R.A.; Maccarrone, M. Resolution of inflammation is altered in chronic heart failure and entails a dysfunctional responsiveness of T lymphocytes. FASEB J. 2018, 33, 909–916.
- Li, T.; Liu, J.; Guo, G.; Ning, B.; Li, X.; Zhu, G.; Yang, D.; Moran, T.H.; Smith, W.W. Synphilin-1 Interacts with AMPK and Increases AMPK Phosphorylation. Int. J. Mol. Sci. 2020, 21, 4352.
- Elesela, S.; Morris, S.B.; Narayanan, S.; Kumar, S.; Lombard, D.B.; Lukacs, N.W. Sirtuin 1 regulates mitochondrial function and immune homeostasis in respiratory syncytial virus infected dendritic cells. PLoS Pathog. 2020, 16, e1008319.
- Mills, C.A.; Trub, A.G.; Hirschey, M.D. Sensing Mitochondrial Acetyl-CoA to Tune Respiration. Trends Endocrinol. Metab. 2018, 30, 1–3.
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. Elife 2019, 8, e47866.
- Sun, H.-X.; Zeng, D.-Y.; Li, R.-T.; Pang, R.-P.; Yang, H.; Hu, Y.-L.; Zhang, Q.; Jiang, Y.; Huang, L.-Y.; Tang, Y.-B.; et al. Essential Role of MicroRNA-155 in Regulating Endothelium-Dependent Vasorelaxation by Targeting Endothelial Nitric Oxide Synthase. Hypertension 2012, 60, 1407–1414.
- Curtis, A.M.; Fagundes, C.T.; Yang, G.; Palsson-McDermott, E.M.; Wochal, P.; McGettrick, A.F.; Foley, N.H.; Early, J.O.; Chen, L.; Zhang, H.; et al. Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proc. Natl. Acad. Sci. USA 2015, 112, 7231–7236.
- Tryggestad, J.B.; Teague, A.M.; Sparling, D.P.; Jiang, S.; Chernausek, S.D. Macrophage-Derived microRNA-155 Increases in Obesity and Influences Adipocyte Metabolism by Targeting Peroxisome Proliferator-Activated Receptor Gamma. Obesity 2019, 27, 1856–1864.
- El Samaloty, N.M.; Hassan, Z.A.; Hefny, Z.; Abdelaziz, D.H.A. Circulating microRNA-155 is associated with insulin resistance in chronic hepatitis C patients. Arab. J. Gastroenterol. 2019, 20, 1–7.
- Hu, J.; Huang, C.-X.; Rao, P.-P.; Cao, G.-Q.; Zhang, Y.; Zhou, J.-P.; Zhu, L.-Y.; Liu, M.-X.; Zhang, G. MicroRNA-155 inhibition attenuates endoplasmic reticulum stress-induced cardiomyocyte apoptosis following myocardial infarction via reducing macrophage inflammation. Eur. J. Pharmacol. 2019, 857, 172449.
- Li, Y.; Tian, Z.; Tan, Y.; Lian, G.; Chen, S.; Chen, S.; Li, J.; Li, X.; Huang, K.; Chen, Y. Bmi-1-induced miR-27a and miR-155 promote tumor metastasis and chemoresistance by targeting RKIP in gastric cancer. Mol. Cancer 2020, 19, 1–14.
- Sun, X.; Song, M.; Song, H.; Wang, Y.; Luo, M.; Yin, L. miR-155 mediates inflammatory injury of hippocampal neuronal cells via the activation of microglia. Mol. Med. Rep. 2019, 19, 2627–2635.
- Ekiz, H.A.; Ramstead, A.G.; Lee, S.-H.; Nelson, M.C.; Bauer, K.M.; Wallace, J.A.; Hu, R.; Round, J.L.; Rutter, J.; Drummond, M.J.; et al. T Cell–Expressed microRNA-155 Reduces Lifespan in a Mouse Model of Age-Related Chronic Inflammation. J. Immunol. 2020, 204, 2064–2075.
- Xia, Y.; Chen, S.; Zeng, S.; Zhao, Y.; Zhu, C.; Deng, B.; Zhu, G.; Yin, J.; Wang, W.; Hardeland, R.; et al. Melatonin in macrophage biology: Current understanding and future perspectives. J. Pineal Res. 2019, 66, e12547.
- He, M.; Xu, Z.; Ding, T.; Kuang, D.M.; Zheng, L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell. Mol. Immunol. 2009, 6, 343–352.
- Ge, J.; Huang, Z.; Liu, H.; Chen, J.; Xie, Z.; Chen, Z.; Peng, J.; Sun, J.; Hou, J.; Zhang, X. Lower Expression of MicroRNA-155 Contributes to Dysfunction of Natural Killer Cells in Patients with Chronic Hepatitis B. Front. Immunol. 2017, 8, 1173.
- Alter, G.; Suscovich, T.J.; Kleyman, M.; Teigen, N.; Streeck, H.; Zaman, M.T.; Meier, A.; Altfeld, M. Low perforin and elevated SHIP-1 expression is associated with functional anergy of natural killer cells in chronic HIV-1 infection. AIDS 2006, 20, 1549–1551.
- Stelekati, E.; Chen, Z.; Manne, S.; Kurachi, M.; Ali, M.-A.; Lewy, K.; Cai, Z.; Nzingha, K.; McLane, L.M.; Hope, J.L.; et al. Long-Term Persistence of Exhausted CD8 T Cells in Chronic Infection Is Regulated by MicroRNA-155. Cell Rep. 2018, 23, 2142–2156.
- Hsin, J.-P.; Lu, Y.; Loeb, G.B.; Leslie, C.S.; Rudensky, A.Y. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat. Immunol. 2018, 19, 1137–1145.
- Qiu, L.-P.; Zhang, Y.; Do, D.C.; Ke, X.; Zhang, S.; Lambert, K.; Kumar, S.; Hu, C.; Zhou, Y.-F.; Ishmael, F.T.; et al. miR-155 Modulates Cockroach Allergen– and Oxidative Stress–Induced Cyclooxygenase-2 in Asthma. J. Immunol. 2018, 201, 916–929.
- Comer, B.S. Does miRNA-155 Promote Cyclooxygenase-2 Expression in Cancer? Drug Dev. Res. 2015, 76, 354–356.
- Souza, P.R.; Marques, R.M.; Gomez, E.A.; Colas, R.A.; De Matteis, R.; Zak, A.; Patel, M.; Collier, D.J.; Dalli, J. Enriched Marine Oil Supplements Increase Peripheral Blood Specialized Pro-Resolving Mediators Concentrations and Reprogram Host Immune Responses. Circ. Res. 2020, 126, 75–90.
- Lannan, K.L.; Spinelli, S.L.; Blumberg, N.; Phipps, R.P. Maresin 1 induces a novel pro-resolving phenotype in human platelets. J. Thromb. Haemost. 2017, 15, 802–813.
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci. Transl. Med. 2013, 5, 174ra26.
- Zhang, L.; Deng, M.; Lu, A.; Chen, Y.; Chen, Y.; Wu, C.; Tan, Z.; Boini, K.M.; Yang, T.; Zhu, Q.; et al. Sodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathway via a HDAC5/HDAC6-dependent mechanism. J. Cell. Mol. Med. 2019, 23, 8139–8150.
- Torun, A.; Enayat, S.; Sheraj, I.; Tunçer, S.; Ülgen, D.H.; Banerjee, S. Butyrate mediated regulation of RNA binding proteins in the post-transcriptional regulation of inflammatory gene expression. Cell. Signal. 2019, 64, 109410.
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism—Metabolites and cofactors. Nat. Rev. Endocrinol. 2015, 12, 43–60.
- Li, N.; Liu, X.-X.; Hong, M.; Huang, X.-Z.; Chen, H.; Xu, J.-H.; Wang, C.; Zhang, Y.-X.; Zhong, J.; Nie, H.; et al. Sodium butyrate alleviates LPS-induced acute lung injury in mice via inhibiting HMGB1 release. Int. Immunopharmacol. 2018, 56, 242–248.
- Huang, Q.; Fei, X.; Li, S.; Xu, C.; Tu, C.; Jiang, L.; Wo, M. Predicting significance of COX-2 expression of peripheral blood monocyte in patients with coronary artery disease. Ann. Transl. Med. 2019, 7, 483.
- Wang, Y.; Xu, Y.; Yang, M.; Zhang, M.; Xiao, M.; Li, X. Butyrate mitigates TNF-α-induced attachment of monocytes to endothelial cells. J. Bioenerg. Biomembr. 2020, 52, 247–256.

