Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jerzy Palka | + 2299 word(s) | 2299 | 2021-12-13 05:02:20 | | | |
| 2 | Yvaine Wei | Meta information modification | 2299 | 2021-12-24 01:53:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Palka, J. Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival. Encyclopedia. Available online: https://encyclopedia.pub/entry/17498 (accessed on 27 July 2026).
Palka J. Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival. Encyclopedia. Available at: https://encyclopedia.pub/entry/17498. Accessed July 27, 2026.
Palka, Jerzy. "Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival" Encyclopedia, https://encyclopedia.pub/entry/17498 (accessed July 27, 2026).
Palka, J. (2021, December 23). Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival. In Encyclopedia. https://encyclopedia.pub/entry/17498
Palka, Jerzy. "Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival." Encyclopedia. Web. 23 December, 2021.
Copy Citation
The estrogen receptor (ER) status and the availability of agonists or antagonists of these receptors determine the processes of growth, differentiation, and proliferation of breast cancer cells. Estrogens and anti-estrogenic compounds have been shown to influence breast cancer cell survival/apoptosis via action through the mitochondrial enzyme proline dehydrogenase/proline oxidase (PRODH/POX).
estrogens
estrogen receptor
breast cancer
1. Introduction
Breast cancer was the most common malignant neoplasm in women and accounted for 11.7% of all cancers globally. WHO cites obesity as one of the main reasons for the high incidence of the disease. The recent increase in the mortality of breast cancers was due to the COVID-19 pandemic that affected both therapy and prevention of the disease [1][2]. Although several therapeutic approaches for breast cancer treatment have been established, the role of estrogen receptor (ER) status in the complex regulatory mechanisms driving apoptosis/survival of cancer cells is not fully understood.
The presence of the ER (ER+) in breast cancers increases positive response to anticancer treatment. Moreover, a better prognosis concerns progesterone receptors (PR+) and human epidermal growth factor (HER2+) positive cancers. The absence of ER is a significant risk factor for relapse and shorter life expectancy. Some authors emphasize that at least a two-receptor ER+PR+HER- expansion profile has a better prognosis than a single-receptor profile such as ER+PR-HER- or ER-PR+HER- [3]. This is probably due to the hormonal reorganization of cellular metabolism driving pro-survival or pro-apoptotic pathways. However, the mechanisms driving apoptosis/survival are not fully understood.
2. Estrogen Receptors Structure, Location and Function
Two distinct estrogen receptor (ER) types, ERα and ERβ, are known to be encoded by two different genes located on two different chromosomes. ERα and ERβ are encoded by ESR1 (chromosome 6, region q24-q27) and ESR2 gene (chromosome 14, region q23.2). The molecular weight of ERα is 67 kDa, the ERβ isoform has 57 kDa [4]. Both types are composed of 6 functional domains named A–F [5]. Domains A and B are located at the amino terminal of the protein. The domain AF1 is able to activate gene transcription in the absence of bound ligand (e.g., the estrogen); however, the activation is weak. Domain C is responsible for receptor dimerization and binding of the ligand-receptor complex to a specific sequence on DNA. The D domain is also called the hinge. It has DNA-binding properties, and its sequence is more variable than that of the C domain. Next is the E domain, which contains a hydrophobic pocket structure called the ligand-binding domain (LBD). The E domain also enables dimerization of nuclear receptors. Some receptors also have an F domain, whose role is not fully elucidated (Figure 1) [5].

Figure 1. The structure of the estrogen receptor. ERα—Estrogen Receptor α; ERβ—Estrogen Receptor β; AF1—activator of transcription 1; C-DBD—DNA Binding Domain, domain C; D-H—Domain D-hinge; E-LBD—Ligand Binding Domain, domain E; AF-2—activator of transcription 2; NH2—amino-terminus, NH2—terminus, N—terminal end or amine-terminus; COOH—carboxylic terminus.
Non-active ERs occur in the cell cytosol, where they form large complexes with chaperone proteins of the HSP (Heat Shock Proteins) family. In this form, they are still inactive but capable of ligand attachment [5]. Ligand binding causes dimerization of the receptor. This process is crucial for the formation of a functional transcription factor and the regulation of gene transcription interacting with the Estrogen Response Element (ERE) (Figure 2). The molecule required for the binding of ER to DNA is FoxA1. It is a critical factor that promotes binding to chromatin [6].

Figure 2. ER-dependent gene transcription. E2—estradiol; ER—Estrogen Receptor, HSP—Heat Shock Proteins; FoxA1—Forkhead box protein A1; ERE—Estrogen Response Element; mRNA—messenger RNA.
The distribution of ERα and ERβ receptors in tissues and organs varies. In most tissues and organs, both types of estrogen receptors are present, while in some, only one type predominates. In the ovaries, uterus, mammary gland, kidney, adrenal gland, testes, epididymis, pituitary gland, and hypothalamus, ERα expression is higher [7][8][9] than in the urinary bladder, prostate gland, heart, and liver [10]. The highest level of ERβ expression was found in the ovary and prostate gland [11]. An important function of estrogen receptors is transcriptional and post-transcriptional regulation of cellular metabolism [12]. It has been suggested that ERα is involved in the regulation of cell proliferation, while ERβ evokes anti-proliferative and pro-apoptotic activity [13][14]. However, ERs comprise also several membranes bound receptors as G protein-coupled estrogen receptor (GPER) and Gq-coupled membrane estrogen receptor (GqmER). Recent studies revealed a functional link between all types of ERs. Interestingly, several oncogenic miRNAs have been shown to modulate the expression of ERs affecting malignant behaviour of cancer cells [15]. Moreover, a ligand-independent signaling has been reported for ERα through kind of cross-talk with epidermal growth factor or insulin-like growth factor-I [16][17]. Whether they are involved in PRODH/POX-dependent regulation of apoptosis/survival requires to be explored.
3. Apoptosis
Apoptosis is the process of programmed cell death, important in the development and homeostasis of multicellular organisms [18]. This process enables the elimination of damaged, old or unnecessary cells. Initiation of the apoptosis pathway is one of the possible cell responses to intracellular or extracellular action of the chemical, physical or biological factors. The external factors that cause cell damage include UV radiation, ionizing radiation, thermal shock, low availability of oxygen and nutrients, drugs, or viral and bacterial infections [19]. The internal factors are activated by oncogenes, cell cycle defects, deficiency of growth factors, energy, hormonal deregulation, etc. [20][21]. Factors inducing apoptosis contribute to the development of neurodegenerative and autoimmune diseases, growth defects, and cancer. The disturbed balance between survival and apoptosis is a common feature of cancer cells [22]. It is also the cause of resistance to chemotherapy, radiotherapy, hormonal and immune therapy [23].
Apoptosis is a precisely regulated process by several classes of proteins. The most important are caspases (a family of intracellular cysteine proteases). They are divided into initiator, implementing, and inflammation caspases. Another important protein in the apoptosis process is the family of BCL-2 proteins (Bax; Bak, Bid, Bim), which have proapoptotic, antiapoptotic, and regulatory activities [24].
Several pathways lead to the induction of apoptosis. The extrinsic pathway is initiated by binding a ligand to the death surface receptors [25]. The intrinsic pathway of apoptosis can be activated by proapoptotic factors released from mitochondria. Apoptogenic molecules that are produced during intracellular stress leads to the increase in permeation of mitochondria. Both pathways stimulate apoptosis through proteolytic cleavage of pro-caspases into active enzymes [26]. The initiator caspases include caspase-8, -9, -10, whereas caspases-3, -6, and -7 are called effector caspases [27]. They can disrupt entire cells within a few minutes.
3.1. The Extrinsic Apoptosis Pathway
The extrinsic process of apoptosis is induced in the cell through the signals from other cells activating the death receptor, which initiates a cascade of intracellular effector proteins [28][29]. Tumor necrosis factor (TNF) is the best-characterized protein that initiates programmed cell death [30]. The same superfamily includes ligand of TNF family receptors (THANK), lymphotoxin (LT), Fas Ligand (FasL), TNF-related apoptosis-inducing ligand (TRAIL), or the Vascular Endothelial Growth Inhibitor (VEGI) [31]. Some of them contain an intracellular death domain (DD). During protein binding to the receptors of the TNF family, the TRADD (Tumor necrosis factor receptor type 1-associated DEATH domain protein) or FADD (Fas-associated protein with death domain) adapter proteins interact with the DD region. Subsequently, the DISC complex (Death-inducing signaling complex) is formed [32][33][34]. This complex combines procaspases -8 and -10 and has autoproteolytic activation properties [35]. Cleaved caspases -8 and -10 activate the implementing caspases and initiate changes in the cell structure leading to cell death [32]. In addition, active caspases -8 and -10 activate BID (a pro-apoptotic BCL family protein), which leads to increased release of cytochrome C from mitochondria by its truncated form tBID (Figure 3).
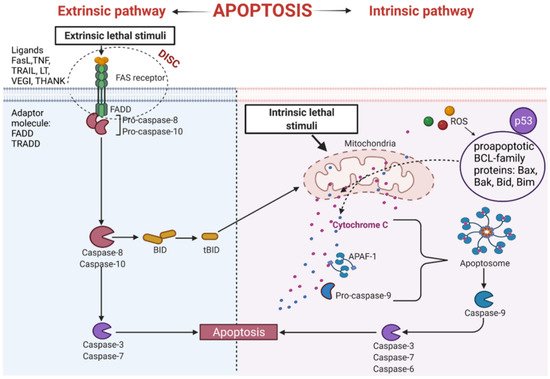
Figure 3. Intrinsic and extrinsic apoptotic pathway. THANK—TNF family receptor; LT—lymphotoxin; FasL—Fas Ligand; TRAIL—TNF-related apoptosis-inducing ligand; VEGI—Vascular Endothelial Growth Inhibitor; TNF—tumor necrosis factor; DISC—Death-inducing signaling complex; FADD—Fas-associated protein with death domain); TRADD—tumor necrosis factor receptor type 1-associated DEATH domain protein; p53—tumor protein p53; APAF-1—apoptotic protease activating factor-1; ROS—reactive oxygen species; Bax, Bak, Bid, Bim—proapoptotic BCL-family proteins; tBID—truncated BID.
An important apoptosis inducer is a p53 protein. This protein participates in the external and internal pathways of apoptosis. p53 interacts with BCL (B-cell lymphoma) proteins family contributing to the upregulation of mitochondrial channels and the cytochrome C efflux into the cytoplasm, activating the internal pathway of programmed cell death [36][37]. It has been established that p53 also induces genes coding for death receptors and death ligands [37].
3.2. The Intrinsic Apoptosis Pathway
This pathway is also called a mitochondrial pathway. It depends on energetic and metabolic processes in the cells and is induced by stress factors. These factors are oxidative stress, DNA damage, changes in cytoplasmic calcium ions concentration, and others. Furthermore, the production of reactive oxygen species (ROS) activates pro-apoptotic BCL- family proteins [38]. As a result of these reactions, the mitochondrial membrane is leaking [39], leading to the release of cytochrome C from mitochondria [38]. Released cytochrome C binds with procaspase9 and apoptotic protease activating factor-1 (APAF-1), forming apoptosome complex. The complex activates the cascade of structural changes in the cell that contribute to cell death through active forms of executive caspases such as caspase-3, caspase-6, and caspase-7 (Figure 3) [40][41].
4. Functional Significance of PRODH/POX in Cell Metabolism
Proline oxidase (POX), also known as proline dehydrogenase (PRODH), is a mitochondrial flavin enzyme associated with the inner mitochondrial membrane. The enzyme catalyzes proline degradation by converting this amino acid to Δ1-pyrroline-5-carboxylic acid (P5C). During this reaction, electrons are transferred via flavin adenine dinucleotide (FAD) to cytochrome C in the respiratory chain, producing ATP molecules, facilitating survival. However, when electrons are transferred directly to oxygen, that happens in specific metabolic conditions, ROS are formed, inducing apoptosis or autophagy [42][43][44][45].
Although the mechanism for switching from ATP to ROS production is not fully understood, it has been suggested that excessive rates of electron transport may contribute to ROS generation [46]. The mechanism of this process is based on mitochondrial membrane potential driving ATP synthase and ATP production and the Kadenbach mechanism (occurring at high ATP/ADP radio) that involves binding of ATP to cytochrome c oxidase (CytOx) and inhibition of the enzyme. In stress situation, ATP-dependent inhibition is switched off and CytOx activity is determined by membrane potential leading to an increase in ROS production. Another mechanism depends on the quantity of electron transfer to the Heme aa3 of CytOx and, in case CytOx is inhibited by ATP, ROS production is decreased. Whether PRODH/POX-dependent ATP/ROS generation involves the same mechanism requires to be explored. However, it has been found that PRODH/POX binds to Coenzyme Q1 (coQ1) decreasing respiratory fitness that was counteracted by N-acetyl-cysteine, suggesting that the effect was mediated by PRODH/POX-dependent ROS formation [47]. Of interest is also finding that PRODH/POX is inhibited by succinate alleviating PRODH/POX effects on respiratory fitness. It suggests that PRODH/POX-induced ATP or ROS formation is metabolic contextdependent.
5. Involvement of ER Agonists in PRODH/POX-Dependent Apoptosis
ERs regulate the expression of AMP kinase (AMPK), which stimulates the activity of PRODH/POX [48][49].
The primary ligands for ER are estrogens, which represent a group of pleiotropic hormones. There are two dominant sources of estrogens in female physiology. In the pre-menopausal age, the ovaries are the principal producer of estrogens. In the post-menopausal age, when ovarian estrogen production declines, fat tissue becomes the main source. Adipocytes have a specific enzyme called aromatase, which converts testosterone to estrogen [50]. ER ligands—estrone, estriol, estradiol, and 2-hydroxy estrone—play functional roles in the physiology of the central nervous system, bones, reproductive and cardiovascular system. However, they also play an important role in carcinogenesis, stimulating cancer cell growth. These hormones act on the cancer cells by targeting the steroid receptor complex to specific DNA sequences, activating specific gene transcription. Several studies have demonstrated this mechanism using tamoxifen, a selective estrogen receptor modulator that inhibits estrogen-dependent tumor growth [51].
Estrogens regulate PRODH/POX-dependent functions at the level of ER, p53, substrate availability for PRODH/POX that is dependent on prolidase activity (proline supporting enzyme) and collagen biosynthesis (proline utilizing process), as well as HIF-1α. It seems that the most important player in determining pro-apoptotic/anti-apoptotic phenotype of cancer cells is the correlation between ERα, P53, and PRODH/POX. As pointed out in the above section, PRODH/POX is a P53-induced gene promoting apoptosis. However, ERα antagonizes P53-dependent apoptosis, promoting cell survival [52][53][54]. Based on these data, it has been established the mechanism for ERα anti-apoptotic potential, suggesting the formation of ERα-P53 complex [55]. Since ERβ was found to attenuate the complex formation, it was concluded that ERβ has pro-apoptotic activity [55]. Whether pro-apoptotic activity of ERβ undergoes through PRODH/POX that has either pro-apoptotic or pro-survival potential requires further study.
6. Effects of ER Modulators on PRODH/POX-Dependent Apoptosis
Phytoestrogens are natural compounds that are ER modulators. They resemble estrogens in their structure. Phytoestrogen’s ability to binding ER induces an estrogenic response or an anti-estrogenic effect [56]. This effect depends on the concentration of the compound and the type of target tissue. Isoflavones at low concentrations have an agonist effect, and at higher concentrations, they are antagonists. Due to this feature, phytoestrogens are called selective estrogen receptor modulators (SERMs) [57]. Phytoestrogens exhibit a broad spectrum of anticancer activity. They inhibit proliferation, invasiveness and induce apoptosis of breast cancer cells. Furthermore, they modulate the activity of ROS-scavenging enzymes [58][59]. For instance, genistein is a characteristic isoflavone found in soybean and is the most abundant natural ERβ modulator. It has an affinity for both ERα and ERβ. However, it has a ninefold preferential affinity for ERβ. By regulating ERβ expression, genistein exerts anticancer effects. Numerous in vitro and in vivo studies have shown that genistein decreases cancer cell proliferation by blocking the cell cycle in the G2/M phase. Induction of apoptosis is associated with the activation of caspase-9 and downregulation of cyclin B1 [60][61].
References
- World Cancer Day: Breast Cancer Overtakes Lung Cancer in Terms of Number of New Cancer Cases Worldwide. IARC Showcases Key Research Projects to Address Breast Cancer; WHO (World Health Organization). Available online: https://www.iarc.who.int/news-events/world-cancer-day-2021 (accessed on 24 November 2021).
- Breast Cancer Now Most Common Form of Cancer: WHO Taking Action. WHO (World Health Organization). Available online: https://www.who.int/news/item/03-02-2021-breast-cancer-now-most-common-form-of-cancer-who-taking-action (accessed on 3 August 2021).
- Shi, J.; Kobayashi, L.C.; Grundy, A.; Richardson, H.; SenGupta, S.K.; Lohrisch, C.A.; Spinelli, J.J.; Aronson, K.J. Lifetime moderate-to-vigorous physical activity and ER/PR/HER-defined post-menopausal breast cancer risk. Breast Cancer Res. Treat. 2017, 165, 201–213.
- Fuller, P.J. The steroid receptor superfamily: Mechanisms of diversity. FASEB J. 1991, 5, 3092–3099.
- Pratt, S.E.; Pollak, M.N. Estrogen and antiestrogen modulation of MCF7 human breast cancer cell proliferation is associated with specific alterations in accumulation of insulin-like growth factor-binding proteins in conditioned media. Cancer Res. 1993, 53, 5193–5198.
- Kumar, U.; Ardasheva, A.; Mahmud, Z.; Coombes, R.C.; Yague, E. FOXA1 is a determinant of drug resistance in breast cancer cells. Breast Cancer Res. Treat. 2021, 186, 317–326.
- Kelly, M.J.; Qiu, J. Estrogen signaling in hypothalamic circuits controlling reproduction. Brain Res. 2010, 1364, 44–52.
- Pelletier, G.; El-Alfy, M. Immunocytochemical localization of estrogen receptors alpha and beta in the human reproductive organs. J. Clin. Endocrinol. Metab. 2000, 85, 4835–4840.
- Friend, K.E.; Chiou, Y.K.; Lopes, M.B.; Laws, E.R., Jr.; Hughes, K.M.; Shupnik, M.A. Estrogen receptor expression in human pituitary: Correlation with immunohistochemistry in normal tissue, and immunohistochemistry and morphology in macroadenomas. J. Clin. Endocrinol. Metab. 1994, 78, 1497–1504.
- Ahlbory-Dieker, D.L.; Stride, B.D.; Leder, G.; Schkoldow, J.; Trolenberg, S.; Seidel, H.; Otto, C.; Sommer, A.; Parker, M.G.; Schutz, G.; et al. DNA binding by estrogen receptor-alpha is essential for the transcriptional response to estrogen in the liver and the uterus. Mol. Endocrinol. 2009, 23, 1544–1555.
- Hiroi, H.; Tsutsumi, O.; Momoeda, M.; Takai, Y.; Osuga, Y.; Taketani, Y. Differential interactions of bisphenol A and 17beta-estradiol with estrogen receptor alpha (ERalpha) and ERbeta. Endocr. J. 1999, 46, 773–778.
- Jensen, E.V.; Jordan, V.C. The estrogen receptor: A model for molecular medicine. Clin. Cancer Res. 2003, 9, 1980–1989.
- Leygue, E.; Murphy, L.C. A bi-faceted role of estrogen receptor β in breast cancer. Endocr.-Relat. Cancer 2013, 20, R127–R139.
- Marino, M.; Ascenzi, P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids 2008, 73, 853–858.
- Taheri, M.; Shoorei, H.; Dinger, M.E.; Ghafouri-Fard, S. Perspectives on the role of non-coding RNAs in the regulation of expression and function of the estrogen receptor. Cancers 2020, 12, 2162.
- Curtis, S.W.; Washburn, T.; Sewall, C.; DiAugustine, R.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630.
- Ignar-Trowbridge, D.M.; Pimentel, M.; Parker, M.G.; McLachlan, J.A.; Korach, K.S. Peptide growth factor cross-talk with the estrogen receptor requires the A/B domain and occurs independently of protein kinase C or estradiol. Endocrinology 1996, 137, 1735–1744.
- Ma, D.; Collins, J.; Hudlicky, T.; Pandey, S. Enhancement of apoptotic and autophagic induction by a novel synthetic C-1 analogue of 7-deoxypancratistatin in human breast adenocarcinoma and neuroblastoma cells with tamoxifen. J. Vis. Exp. 2012, 30, 3586.
- Sen, S. Programmed cell death: Concept, mechanism and control. Biol. Rev. Camb. Philos. Soc. 1992, 67, 287–319.
- Parton, M.; Dowsett, M.; Smith, I. Studies of apoptosis in breast cancer. BMJ Open 2001, 322, 1528–1532.
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430.
- Shi, Y. Apoptosome: The cellular engine for the activation of caspase-9. Structure 2002, 10, 285–288.
- Reed, J.C. Apoptosis mechanisms: Implications for cancer drug discovery. Oncology 2004, 18, 11–20.
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027.
- Eissing, T.; Conzelmann, H.; Gilles, E.D.; Allgower, F.; Bullinger, E.; Scheurich, P. Bistability analyses of a caspase activation model for receptor-induced apoptosis. J. Biol. Chem. 2004, 279, 36892–36897.
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316.
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189.
- Ashkenazi, A.; Dixit, V.M. Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol. 1999, 11, 255–260.
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308.
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354.
- Piotrowska, A.; Izykowska, I.; Podhorska-Okolow, M.; Zabel, M.; Dziegiel, P. The structure of NF- kappaB family proteins and their role in apoptosis. Postep. Hig. Med. Dosw. 2008, 62, 64–74.
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302.
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis-the p53 network. J. Cell Sci. 2003, 116, 4077–4085.
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756.
- Peter, M.E.; Krammer, P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003, 10, 26–35.
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694.
- Wu, G.S.; Kim, K.; el-Deiry, W.S. KILLER/DR5, a novel DNA-damage inducible death receptor gene, links the p53-tumor suppressor to caspase activation and apoptotic death. Adv. Exp. Med. Biol. 2000, 465, 143–151.
- Daniel, P.T. Dissecting the pathways to death. Leukemia 2000, 14, 2035–2044.
- Jiang, P.; Gan, M.; Lin, W.L.; Yen, S.H. Nutrient deprivation induces alpha-synuclein aggregation through endoplasmic reticulum stress response and SREBP2 pathway. Front. Aging Neurosci. 2014, 6, 268.
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896.
- Schattenberg, J.M.; Galle, P.R.; Schuchmann, M. Apoptosis in liver disease. Liver Int. 2006, 26, 904–911.
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015, 356, 165–170.
- Phang, J.M.; Donald, S.P.; Pandhare, J.; Liu, Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 2008, 35, 681–690.
- Arentson, B.W.; Sanyal, N.; Becker, D.F. Substrate channeling in proline metabolism. Front. Biosci. 2012, 17, 375–388.
- Pandhare, J.; Donald, S.P.; Cooper, S.K.; Phang, J.M. Regulation and function of proline oxidase under nutrient stress. J. Cell. Biochem. 2009, 107, 759–768.
- Vogt, S.; Rhiel, A.; Weber, P.; Ramzan, R. Revisiting Kadenbach: Electron flux rate through cytochrome c-oxidase determines the ATP-inhibitory effect and subsequent production of ROS. Bioessays 2016, 38, 556–567.
- Hancock, C.N.; Liu, W.; Alvord, W.G.; Phang, J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids 2016, 48, 859–872.
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J.; et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559.
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 2012, 8, 1407–1409.
- Di Nardo, G.; Zhang, C.; Marcelli, A.G.; Gilardi, G. Molecular and structural evolution of cytochrome P450 aromatase. Int. J. Mol. Sci. 2021, 22, 631.
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618.
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065.
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A.; et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086.
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840.
- Lu, W.; Katzenellenbogen, B.S. Estrogen receptor-beta modulation of the ERalpha-p53 loop regulating gene expression, proliferation, and apoptosis in breast cancer. Horm. Cancer 2017, 8, 230–242.
- Younes, M.; Honma, N. Estrogen receptor beta. Arch. Pathol. Lab. Med. 2011, 135, 63–66.
- Messina, M. Soy and health update: Evaluation of the clinical and epidemiologic literature. Nutrients 2016, 8, 754.
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients 2018, 10, 1618.
- Rodriguez-Garcia, C.; Sanchez-Quesada, C.; Gaforio, J.J. Dietary flavonoids as cancer chemopreventive agents: An updated review of human studies. Antioxidants 2019, 8, 137.
- Chirumbolo, S.; Bjorklund, G.; Lysiuk, R.; Vella, A.; Lenchyk, L.; Upyr, T. Targeting cancer with phytochemicals via their fine tuning of the cell survival signaling pathways. Int. J. Mol. Sci. 2018, 19, 3568.
- Perez-Vizcaino, F.; Fraga, C.G. Research trends in flavonoids and health. Arch. Biochem. Biophys. 2018, 646, 107–112.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
817
Revisions:
2 times
(View History)
Update Date:
29 Mar 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No