Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Davide Marchi | + 4476 word(s) | 4476 | 2021-12-21 07:27:34 | | | |
| 2 | Camila Xu | Meta information modification | 4476 | 2021-12-23 02:34:23 | | | | |
| 3 | Camila Xu | Meta information modification | 4476 | 2021-12-23 02:59:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Marchi, D. Glucocorticoid Receptor Activity. Encyclopedia. Available online: https://encyclopedia.pub/entry/17452 (accessed on 26 June 2026).
Marchi D. Glucocorticoid Receptor Activity. Encyclopedia. Available at: https://encyclopedia.pub/entry/17452. Accessed June 26, 2026.
Marchi, Davide. "Glucocorticoid Receptor Activity" Encyclopedia, https://encyclopedia.pub/entry/17452 (accessed June 26, 2026).
Marchi, D. (2021, December 22). Glucocorticoid Receptor Activity. In Encyclopedia. https://encyclopedia.pub/entry/17452
Marchi, Davide. "Glucocorticoid Receptor Activity." Encyclopedia. Web. 22 December, 2021.
Copy Citation
Glucocorticoids (GCs) represent a well-known class of lipophilic steroid hormones biosynthesised, with a circadian rhythm, by the adrenal glands in humans and by the inter-renal tissue in teleost fish (e.g., zebrafish).
glucocorticoid
glucocorticoid receptor
hypoxia inducible factor
crosstalk
inflammation
immune modulations
1. Introduction
The name “glucocorticoid” (GC) is a portmanteau word (glucose + cortex + steroid), which derives from their key role in the regulation of glucose metabolism, their biosynthesis at the level of the adrenal cortex and their steroidal structure. They represent a well-known class of lipophilic steroid hormones synthetized, with a circadian rhythm, by the adrenal glands in humans and by the inter-renal tissue in teleost fish. GC circadian production in mammals is tuned by the hypothalamus-pituitary-adrenal (HPA) axis, which is the equivalent of the hypothalamus-pituitary-inter-renal (HPI) axis in teleost fish. Both are essential for stress adaptation [1][2][3][4]. The axis consists of a highly conserved regulatory system present in all living organisms aimed at maintaining a dynamic equilibrium in the body in response to external and internal stimuli, which is fundamental to assure homeostasis and survival. Cortisol, the end-product of the HPA/I axis, is the main GC both in humans and teleost fish and plays a fundamental role in the maintenance of both resting and stress-related responses [5][6][7][8].
Since their discovery in the 1940s [9], much has been learnt of GC molecular modes of action [10][11][12][13][14][15][16][17][18][19][20][21][22][23]. In particular, the glucocorticoid receptor’s characterization as a DNA-binding protein that regulates transcription initiation [24], the cloning of GR [25][26] and the breakthrough that most of the immunosuppressive actions of GCs occur via interfering with key inflammatory transcriptional regulators such as NF-κB and AP-1 [27][28][29][30], represent the main milestones.
Natural and synthetic glucocorticoids have been widely used for decades as effective anti-inflammatory and immunosuppressive treatments to control pathological disorders, which are very often linked to hypoxia. In particular, they have been broadly used to treat both acute and chronic inflammations, including inflammatory bowel disease, rheumatoid arthritis, multiple sclerosis, eczema and psoriasis, as well as being used in treatment of various leukaemias and in immunosuppressive regimes upon organ transplant [31][32][33][34][35][36]. At any point in time, an estimated ~1% of the total adult UK population receives oral glucocorticoid therapy [37]. However, due to the presence of adverse effects [38] and GC resistance [39][40][41][42], their therapeutic benefits are limited in patients chronically treated with these steroids. Examples of the most common GC-related side effects include osteoporosis, glaucoma, diabetes, skin atrophy, abdominal obesity, dyslipidemia, hypertension in adults and growth retardation in children [16][43][44].
Cortisol exerts its functions through direct binding to the glucocorticoid receptor (GR), but also to the mineralocorticoid receptor (MR), which binds cortisol with even higher affinity [45][46]. As transcription factors, both GR and MR compete for the same ligands, can form heterodimers and homodimers with each other, recognize and bind many of the same hormone response elements on the DNA, and share numerous coregulatory proteins involved in the gene transcription initiation. Importantly, GCs activate MR in most tissues at basal levels, whereas activate GR under stressful conditions or at the diurnal peak [47].
Once bound together, they form an active complex which can function in the nucleus to modulate the transcription of effector proteins, as well as in the cytoplasm to hamper targeted transcription factors activity. Historically, these functions have been coined genomic and non-genomic modes of action, respectively [48][49][50][51]. Importantly, GCs and their kindred intracellular receptors, represent critical checkpoints in the endocrine control of vertebrate energy homeostasis. Indeed, if HPA axis activity is not accurately regulated, GC imbalance may result in different pathological conditions such as hypertension, severe cardiovascular, immunological and metabolic complications (e.g., Addison’s disease (GC deficiency) and Cushing’s syndrome (GC excess)) [52][53][54]. In addition, alterations or flaws in the HPA axis response are tightly associated with a broad range of inflammatory and autoimmune diseases, both in humans and in animal models. The latter include Crohn’s disease, rheumatoid arthritis, colitis, inflammatory bowel disease, multiple sclerosis (whose animal equivalent is autoimmune encephalomyelitis), dermatitis, and asthma. Inflammatory conditions include fibromyalgia, chronic fatigue syndrome, depression, and post-traumatic stress disorder (PTSD) [55][56][57][58][59][60][61]. Moreover, even if the biological effects induced by GCs are usually adaptive, their abnormal activity may contribute to a series of acute metabolic diseases which include insulin resistance, obesity, and type 2 diabetes [62][63]. Thus, furthering the research on how GCs precisely work and interact with other pathways may provide better tools to treat these diseases and simultaneously allow the development of selective GR agonists and specific drug-targeting strategies.
Similarly to GCs that are involved in numerous homeostatic maintenance activities (e.g., metabolism of protein, carbohydrate and lipid, etc.) [64], the HIF signalling pathway exerts a pivotal role in ensuring homeostasis, the preservation of which is essential for the correct functioning of the cell. In this regard, the ability to perceive and quickly respond to changes related to environmental oxygen availability is controlled by the hypoxia-inducible factor transcription factors (HIF) family. Hypoxia is a common pathophysiological occurrence, with a profound impact both on human and animal physiology, in which oxygen availability to cells, tissues or to an organ is reduced below a certain threshold (O2 levels < 2%) [65][66]. HIF transcription factors are key homeostatic regulators which coordinate a metabolic shift from aerobic to anaerobic metabolism to assure cell survival, both in mammals and in zebrafish [67][68][69][70][71].
The HIF pathway is finely regulated by the PHD3-VHL-E3 ubiquitin ligase complex the aim of which is to maintain low basal HIF levels that can rapidly increase to promptly respond when oxygen levels decrease. This avoids any activation of the HIF pathway under normoxic conditions. As a transcription factor, HIF drives the hypoxic response via binding to specific hypoxia-response elements (HREs). These are involved in decreasing oxygen consumption and increasing oxygen and nutrient delivery [72][73][74]. Interestingly, HIF signalling can tune its own activation via negative feedback by inducing the expression of the oxygen sensors proteins (PHDs), in particular prolyl hydroxylase 3 (PHD3) in zebrafish and PHD2 in humans and mice [75][76].
However, although the HIF response is aimed at restoring tissue oxygenation and perfusion, it may sometimes be maladaptive and may contribute to the onset of different pathological conditions (e.g., inflammation, stroke, tissue ischemia and growth of solid tumours) [65]. Thus, both glucocorticoids and hypoxia-induced transcriptional responses have been shown to exert crucial roles in tissue homeostasis and in the regulation of cellular responses to stress and inflammation [77][78][79][80][81][82].
2. Glucocorticoids
2.1. Biosynthesis, Secretion and Availability
GCs are essential steroid hormones biosynthesized and secreted by the adrenal cortex/inter-renal gland both in a circadian manner and in response to stress. The latter is generally defined as a status of real or perceived threat to homeostasis. Assuring homeostasis in the presence of stressors requires the activation of an intricate series of coordinated biological responses performed by the nervous, endocrine and immune systems [62][83]. The key anatomical structures that regulate the stress response are located both in the central nervous system and in peripheral tissues. The primary effectors of the stress response are localized in the paraventricular nucleus (PVN) of the hypothalamus, in the anterior lobe of the pituitary gland and at the level of the adrenal gland. These three main structures are generally referred to as the hypothalamic-pituitary-adrenal (HPA) axis in humans, and as the hypothalamic-pituitary-inter-renal axis (HPI) in zebrafish [62][83][84]. Among these, the hypothalamus is the initial stressor recognition site for both internal and external signals. In mammals, neurons localized in the paraventricular nucleus synthesize both corticotropin-releasing factor (CRF) and arginine vasopressin (AVP), which are released into hypophyseal portal vessels that access the anterior pituitary gland. On the other hand, in teleosts, there is a direct neuronal connection to endocrine cells through the hypophyseal stalk, since they lack a portal system between the hypothalamus and the pituitary gland [85]. Here, CRF binding to its receptor localized on pituitary corticotropes triggers the release of adrenocorticotropic hormone (ACTH) into the systemic circulation. In humans, ACTH derives by post translational modification of its precursor encoded by the proopiomelanocortin (POMC) gene. Of note, due to genome duplication, two pomc genes named pomca and pomcb have been identified in zebrafish [86][87][88]. However, only pomca seems to be expressed in the pituitary gland and is required for the inter-renal organ development [89][90]. The main target of ACTH is the adrenal cortex in humans and the inter-renal tissue in teleosts, where it binds to the melanocortin 2 receptor (MC2R) on the steroidogenic cells. Here, it stimulates cortisol biosynthesis and secretion starting from cholesterol [62][83][91][92].
Finally, once released into the systemic circulation, GCs can access target tissues (e.g., liver, heart, and vascular tissues) to exert metabolic and cardiovascular effects and the brain itself, in order to support cognitive processes required to tackle a threatening situation [93]. Under non stressful conditions, glucocorticoid levels in the serum are homeostatically controlled by the HPA monitoring activity, whereas glucocorticoid availability is further tuned at a tissue and cellular level. Circulating glucocorticoids are primarily bound to corticosteroid binding globulin (CBG) and just a small percentage (5–15%) is bound to albumin. As a result, the majority of GC is maintained in an inactive form, and only the remaining 5% of systemic GC is free and bioactive. Hence, CBG concentration constitutes a pivotal regulator of cortisol accessibility [94].
Importantly, GCs are also able to control their own biosynthesis and secretion by tuning the activity of the HPA/I axis itself. This is particularly important to stop the stress response and avoid an exacerbated reaction [95]. This is achieved via a GC-GR mediated negative feedback loop, which acts both at the hypothalamic and anterior pituitary levels, where GC-GR activity inhibits both CRH and POMC (ACTH precursor) biosynthesis and release [2][21][23][96]. This occurs via a mechanism that requires GC-GR binding to an nGRE within the pomca promoter [97]. For these reasons, pomca is a well-established and frequently used readout of GR activity. In addition, GCs may indirectly control the HPA axis activity through modulation of brain structures activity, such as the amygdala, the hippocampus and the prefrontal cortex, that can, in turn, influence the activity of the paraventricular nucleus [93][98][99][100].
2.2. The Glucocorticoid Receptor: Structure and Functions
GCs exert their systemic functions by binding to the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR). Due to their lipophilic nature, GCs can passively diffuse across the plasma membrane into the cytoplasm. Within the cells, their biological availability is then regulated by two enzymes of the 11β-Hydroxysteroid dehydrogenase (11β-HSD) family that work in an opposite fashion. 11β-HSD2 oxidizes cortisol into its inactive form cortisone, reducing GC availability. Vice versa, 11β-HSD1 transforms cortisone to cortisol, thereby increasing local GC activity. Inside the cell, GCs can bind to their specific receptors GR and MR [33][93][101][102]. Both receptors, in the absence of their ligands (unbound state) are associated in an inactive oligomeric complex with specific regulatory proteins. Among these, heat shock protein-90 kD (HSP90), which binds both GR and MR to the C-terminal domain, heat shock protein-70 kD (HSP70), p59 immunophilin, Fkbp51 and Fkbp52 and the small p23 phosphoprotein maintain correct protein folding of the receptor [93][103][104]. The GR, which belongs to the nuclear receptor transcription factor family, is composed of different conserved structural domains [105]. These include an N-terminal variable region (NTD) required for ligand-independent gene transactivation, which contains a transactivation domain named activation function 1 (AF1). The latter is responsible for the transcriptional activation and is involved in the association with coregulators and the basal transcription machinery. A central DNA-binding domain composed of two zinc fingers has been shown to be crucial both for GR homodimerization and DNA-binding specificity. This is followed by an adjacent flexible hinge region allowing proper DNA binding, dimerization, and nuclear translocation of the receptor [106]. Finally, the C-terminal region (LBD) contains the ligand binding domain and a secondary transactivation domain (AF2), regulated by hormone binding, which is essential for dimerization, interaction with cochaperones, coregulators, and other transcription factors [107][108]. The LBD also comprises a dimer interface which is fundamental for GR function and the binding of the heat shock protein 90 (Hsp 90) [109]. Both DBD and LBD include nuclear localization signals, which are required for GR nuclear translocation. Finally, DBD also incorporate the nuclear export signal sequence (NES) which targets it for export from the nucleus to the cytoplasm via the nuclear pore complex [104] (Figure 1).

Figure 1. Glucocorticoid receptor domain structure and translational isoforms. The N-terminal domain (NTD), which is required for ligand-independent gene transactivation, includes a transcriptional activation function region (AF1). The latter, which interacts with coregulators and with the basal transcriptional machinery, is the main posttranslational modifications site. The LBD, which is made up of 12 α-helices and 4 β-sheets, forms a hydrophobic pocket needed for GC binding and includes an AF2 domain. The latter allows interaction with coregulators in a ligand-dependent way. Finally, two nuclear localization signals, named NL1 and NL2, are localized in the DBD-hinge region junction and within the LBD, respectively. Asterisks indicate the location of the starting amino acid (aa position: 1, 27, 86, 90, 98, 316, 331, 336) of the eight different GRα translational isoforms, which are characterised by progressively shorter NTDs.
Although the NTD is conserved, literature reviews and sequence alignments of human, monkey, rat, and mouse GRs have revealed that there are another eight conserved AUG start codons in the exon 2 (Figure 1). In humans, these were shown to produce various GR isoforms with progressively shorter N-terminal transactivation domains [104]. These are formed due to the presence of alternative Kozak translation initiation sequences which can cause either ribosomal shunting or ribosomal leaky scanning mechanisms. This allows the generation of different GR subtypes with truncated N-termini [110][111][112][113][114], which are likely to be fully active. This is consistent with data from zebrafish, where a GR mutant line (grsh551), characterized by a 1 bp deletion in the first coding exon (exon 2, Q48fsX3), proved not to have any detectable phenotype [115]. This was confirmed further by ISH analysis, which showed that both grsh551 mutants and wildtypes displayed an identical downregulated pomca expression after synthetic GC (Betamethasone 17,21-dipropionate) administration.
Synthetic GR agonists are supposed to trigger a potent GC response, which in turn elicits the GC-GR mediated negative feedback loop, aimed to shut down their own biosynthesis. As previously mentioned, this mainly occurs at the level of the pituitary gland via downregulation of pomca [2][97][102]. For this reason, if GR is not functional, the GC-GR negative feedback loop cannot occur and pomca expression should not be downregulated, as occurs in grsh551 mutants. Indeed, in grsh551 mutants the feedback occurs normally.
In addition to alternative starts, alternative splicing at exon 9 is responsible for generating two different GR splice variants, namely GRα (777 aa) and GRβ (742 aa) [25][116]. These two receptor isoforms share an identical amino acid sequence between 1–727 aa and then diverge. In particular, the human (h) GRα c C-terminal region contains 50 distinct amino acid residues that form two alpha-helical structures that play a key ligand binding role. In contrast, the hGRβ C-terminal is characterized by a shortened 15 non-homologous, specific amino acid sequence that prevents GC binding [20][117][118]. Hence, hGRβ does not bind traditional glucocorticoid agonists and lacks transactivational activity on GRE-containing promoters, whereas hGRα is the canonical GR isoform. Nevertheless, hGRβ is constitutively present in the nucleus where it has been shown to act as a dominant-negative inhibitor of hGRα’s transactivational properties [118][119][120]. The mechanism behind this inhibition is still uncertain, but several studies have suggested that competition between both hGR isoforms for transcriptional coactivator proteins and/or the formation of inactive GRα-GRβ heterodimers might be responsible for that [117][121][122][123].
Moreover, the function of hGRβ extends beyond antagonism of the hGRα isoform [124]; for instance, binding to the glucocorticoid antagonist mifepristone (RU486) has been also reported [125][126]. In addition, it has been shown that increased hGRβ expression is correlated both with the development of immune-related diseases (e.g., ulcerative colitis, leukemia and severe asthma) [127][128][129] and with glucocorticoid resistance in patients affected by these diseases [130][131][132]. Interestingly, previous studies have established the occurrence of a GR β-isoform in zebrafish larvae, which similarly to the hGRβ, confirmed the lack of a role in transcriptional regulation and a dominant-negative inhibitor activity on zGRα [19][20][133][134]. In this respect, zebrafish have been shown to be a reliable and useful model system both for GC resistance and glucocorticoid receptor research [2][135].
2.3. GCs Mechanisms of Action
The conventional view of GC mechanism of action has been recently revised and described as a more complicated multiprotein-regulated process. In this regard, it has been shown that in both humans and zebrafish, upon cortisol binding, GR undergoes a conformational change that involves an FKBP51-FKBP52 exchange. The latter triggers the translocation of the GC-GR active complex into the nucleus. FKBP51 is a cochaperone protein that binds HSP90 and decreases the affinity of GR for cortisol. For this reason, FKBP51 has been considered an inhibitor of GR transcriptional activity and its overexpression has been linked to GC resistance in autoimmune diseases [136][137][138]. After ligand binding, FKBP51 is replaced by FKBP52, which in turn recruits dynein to support translocation of the GC/GR complex to the nucleus [139][140]. This structural modification exposes the two GR nuclear localisation signals, allowing the hormone-activated GR to dimerize with another GC-GR molecule and to migrate into the nucleus via nuclear pores [141][142]. Interestingly, this transcription factor complex can also act nongenomically (in the cytoplasm), where it may interact via direct protein-protein interactions with other transcriptional regulators and/or kinases (e.g., basal transcription machinery (BTM); phosphoinositide 3-kinase (PI3K); signal transducer and activator of transcription (STAT)) [63][143][144][145] (Figure 2).
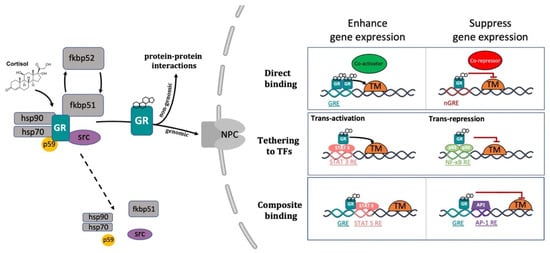
Figure 2. Representative picture of the canonical GR signalling pathway. After binding to GC, GR undergoes a FKBP51-FKBP52 mediated conformational change, becomes hyper-phosphorylated, dissociates from accessory proteins (chaperone complex) and finally translocates into the nucleus. Here, after dimerization with other GRs, it regulates the transcription of target genes by binding to DNA. Interestingly, GR may enhance or repress transcription of target genes by directly binding to palindromic GC response elements (GRE), or by tethering itself to other transcription factors apart from DNA binding, or in a composite manner by both directly binding GRE and interacting with transcription factors bound to neighbouring sites. Created with BioRender.com.
Inside the cell nucleus, GC-GR complexes can directly bind to specific GREs, as tetramers, to upregulate (transactivation) or downregulate (transrepression) the transcription of target genes. Generally, the preferred GRE motif (AGAACAnnnTGTTCT in humans, and GGAACAnnnTGTTCT in zebrafish) is an imperfect palindromic consensus sequence that consists of two 6 bp half sites. The three-nucleotide spacing in-between the two half sites is essential for the GR to tetramerize on this sequence. Previous genome-wide studies have shown that the same GRE can mediate both the GC-dependent induction of many genes (positive GRE) and the repression of others (negative GRE) [104][146]. Interestingly, the presence of specific inverted repeats negative GREs (IR nGRE), unrelated to simple GREs has also been reported both in mice and in humans. These DNA binding sequences are palindromic sequences consisting of two inverted repeated (IR) motifs separated by 1 bp. In particular, they bind GC-GR complexes to promote the assembly of cis-acting GR-SMRT/NCoR repressing complexes [147][148] (Figure 2).
In summary, these findings indicate that the broadly different GC effects on various tissues can be partially ascribed to cell type-specific differences in the chromatin landscape that affects the accessibility of specific GREs for GR binding [149][150]. Furthermore, the GC concentration at which the GR binds to GREs depends on the cell type and chromosomal context. Another important feature of the GC-GR complex that makes its effects even more versatile is that it can tune gene expression in different ways: by binding directly to DNA, by tethering itself to other transcription factors bound to DNA, or via direct binding to DNA and with neighbouring DNA-bound transcriptional regulators (composite manner, Figure 2) [104][151][152].
2.4. The Mineralocorticoid Receptor: Structure and Functions
As previously mentioned, cortisol can bind not only to GR, but also to MR. Both are members of the steroid receptor superfamily (corticosteroid receptors) of ligand-activated transcription factors that enhance or repress the transcription of target genes, as well as promote rapid nongenomic/extra-nuclear events via several cell signalling pathways [47]. The mineralocorticoid receptor is a 984-amino acid cytoplasmic protein that is characterized by three different domains: an N-terminal transcriptional regulator domain, a DNA-binding domain, and a ligand-binding domain responsible for the selectivity of hormone binding. Analogously to GR, in its unliganded state MR is associated with a number of chaperone proteins (HSP90, HSP70, FKBP51 and p23) that play a crucial role in trafficking and maintaining MR in a suitable conformation for ligand binding [153].
MR has a ten-fold higher affinity for cortisol than GR and is preferably activated under basal conditions, implying distinct roles for each receptor in the regulation of HPA axis activity [154][155] It has also been observed that cortisol, even at lower concentrations than those required to activate the GR, binds to MR and might enhance the activity of several kinases (i.e., protein kinase C (PKC), cyclic adenosine 3′,5′-monophosphate (cAMP), and phosphoinositide 3-kinases (PI3K)) involved in different signal transduction cascades [4][47][156]. On the other hand, GR is primarily activated as a result of stress, or at the diurnal peak when circulating cortisol levels are also peaking [154][157]. Cortisol also exerts broad effects on mood and behaviour via MRs and GRs that are expressed in different regions of the brain [158][159]. In particular, GR is widely expressed throughout the brain, primarily in the PVN (stress-regulating centre) and in the prefrontal cortex-hippocampal-amygdala circuitry (cognitive, emotional regulation and memory consolidation centre). Vice versa, MR is predominantly expressed in the hippocampus, amygdala, and the lateral septum (emotionality, social behaviour and feeding process hub) [160].
In mammals, the mineralocorticoid system is essential to regulate potassium and fluid homeostasis upon aldosterone activation of MR. Even though cortisol is a high-affinity ligand for MR, this steroid is deactivated in specific mineralocorticoid responsive tissues, such as the kidney, by the previously mentioned 11b-HSD-2 enzymatic activity. This allows aldosterone, a second corticosteroid present in mammals, to bind to this receptor. Surprisingly, teleosts do not synthesize aldosterone, and cortisol has been shown to mediate stress axis regulation, as well as the majority of the changes in iono-regulatory and osmo-regulatory functions, via GR and MR signalling [1][4].
Interestingly, previous work performed both in rats and in teleosts showed that while MR is involved both in basal and onset of stress-induced HPA/I axis activity, GR mainly controls its termination [4][161]. However, a recent zebrafish study highlighted that rapid locomotor responses to quick changes in light illumination or water salinity (environmental stressors) require GC-GR mediated HPI axis signalling, but not MR [162]. Finally, our recently published research suggested that not only GR, but also MR signalling, is involved in the GC-negative feedback regulation (HPI axis termination) and plays a key role in assuring a proper HIF response in teleosts [163]. In view of this, furthering the precise role of MR and mineralocorticoid modes of action in vivo, particularly in relation to in the HIF signalling pathway, is warranted.
2.5. The Role of Glucocorticoids in Inflammation
Glucocorticoids suppress most of the events early in the inflammatory response and subsequently facilitate inflammation resolution. They suppress both vasodilation and the enhanced vascular permeability that occurs as a consequence of an inflammatory challenge, and inhibit leukocyte migration from the inflamed region [10][164][165]. In addition, they tune both distribution and trafficking of leukocytes, promote death/survival in some cells and may affect cellular differentiation programmes [165][166][167][168][169][170]. It is well established that many of the anti-inflammatory and immunosuppressive glucocorticoid actions are referable either indirectly or directly to GC-GR mediated transcriptional regulation of numerous genes expressed in leukocytes [165]. On the other hand, even though MR expression has been reported in immune cells [171][172][173], its anti-inflammatory role has been considered negligible so far [173][174], but rather surprisingly, MR-dependent proinflammatory effects have also been noted [175][176]. It is presently unclear whether these effects are mediated by the glucocorticoid cortisol or the mineralocorticoid aldosterone [177].
Glucocorticoids, through GR, can modulate gene expression in different ways [103][108]. Among these, transactivation is the mechanism by which GC predominantly induce the transcription of numerous anti-inflammatory genes, such as GILZ, MKP-1 and IĸBα. This occurs through direct binding of single or multiple GC-GRs to palindromic glucocorticoid response elements (GREs) [178][179][180][181]. Importantly, this mode of action has also been shown to be responsible for several undesirable metabolic side effects linked to chronic GC treatment [140]. On the other hand, transrepression is the mechanism by which GC downregulate the transcription of inflammatory genes and requires direct protein-protein interaction of GR to other transcription factors. This mode of action is generally accepted to convey the beneficial GC anti-inflammatory effects, which are mainly implicated in rapid cellular responses [40][140][182][183][184]. In particular, transrepression is known to mainly occur via direct binding between the monomeric GC-GR complex and transcription factors (e.g., AP-1, NF-kB, c-Jun, and c-Fos) activated by cytokines and other pro-inflammatory stimuli, which synergistically coordinate the expression of several proinflammatory genes [27][100][152][185][186][187][188]. Of note, most of these genes are commonly overexpressed during chronic nonresolving inflammatory states. Interestingly, transrepression is not restricted to these transcription factors but also includes others such as CREB, STAT, and T-bet [152][189]. As a result, the mutual antagonism between transcription factors frequently impairs their transcriptional properties and prevents them from binding to their corresponding DNA response elements.
As a consequence of the above considerations, optimal GC analogs should be characterized by a high inhibitory activity against inflammatory mediators, coupled with a low transactivation activity, in order to induce minimal side effects. Interestingly, different steroidal and nonsteroidal ligands have been reported to have this dual function (e.g., RU-24858 and ZK-216348) [140][183][190][191][192]. These compounds have been reported to suppress key inflammatory and immune transcription factor activity in vivo [190][191][192][193][194]. However, as stated before, since GCs can trigger gene expression via multiple routes, unexpected secondary side effects might occur. For this reason, further research is also warranted to elucidate the implications of the nongenomic GC-mediated activity both in the immune and inflammatory scenario.
Importantly, although the GC-GR complex is known for its anti-inflammatory effects, the picture is more complex. Indeed, contrary to expectations, GR loss of function was reported by Facchinello and coworkers to prevent the transcriptional activity linked to the inflammatory immune response (i.e., of cytokines Il6, Il1β, Il8 and Mmp-13) [8], corroborating the hypothesis of a GC-GR mediated dual action on the immune system [35][195]. However, it is clear that further research is warranted to better elucidate this aspect. In addition, GR was shown to synergistically induce proinflammatory genes by acting on other signalling pathways [196][197][198][199]. Finally, studies also demonstrated that GCs increase the transcription of numerous anti-inflammatory molecules such as interleukin-10 (IL-10), interleukin-1 receptor antagonist (IL-1RA), secretory leukocyte inhibitory protein and neutral endopeptidase [41][200].
Previous research also revealed that alterations in chromatin structure are important for modulating the outcome of GC activity. Indeed, GR can interact differently with histone acetyltransferases (HATs), with histone deacetylases (HDACs) and also kinases (i.e., MSK1, PKA and JNK) [152]. These may, in turn, modulate the chromatin environment by modifying chromatin accessibility and further tuning inflammatory and immune gene expression [152]. Furthermore, since chromatin accessibility can predetermine GR binding patterns and is crucial for cell-specific outcomes, it can provide novel molecular basis for tissue selectivity [201][202]. In addition, another study showed that GR may directly inhibit CREB binding protein (CBP)-associated HAT activity and may recruit HDAC2 to the p65-CBP HAT complex [203]. This novel glucocorticoid repression mechanism suggests that histone acetylation inhibition represents an additional level of control of inflammatory gene expression. Consequently, this further indicates that pharmacologically manipulating specific histone acetylation status could be an alternative approach for treating inflammatory diseases.
References
- Alsop, D.; Vijayan, M. The zebrafish stress axis: Molecular fallout from the teleost-specific genome duplication event. Gen. Comp. Endocrinol. 2009, 161, 62–66.
- Griffiths, B.B.; Schoonheim, P.J.; Ziv, L.; Voelker, L.; Baier, H.; Gahtan, E. A zebrafish model of glucocorticoid resistance shows serotonergic modulation of the stress response. Front. Behav. Neurosci. 2012, 6, 68.
- Tokarz, J.; Möller, G.; de Angelis, M.H.; Adamski, J. Zebrafish and steroids: What do we know and what do we need to know? J. Steroid Biochem. Mol. Biol. 2013, 137, 165–173.
- Faught, E.; Vijayan, M.M. The mineralocorticoid receptor is essential for stress axis regulation in zebrafish larvae. Sci. Rep. 2018, 8, 18081.
- Munck, A.; Guyre, P.M.; Holbrook, N.J. Physiological Functions of Glucocorticoids in Stress and Their Relation to Pharmacological Actions. Endocr. Rev. 1984, 5, 25–44.
- Macfarlane, D.P.; Forbes, S.; Walker, B.R. Glucocorticoids and fatty acid metabolism in humans: Fuelling fat redistribution in the metabolic syndrome. J. Endocrinol. 2008, 197, 189–204.
- Kuo, T.; McQueen, A.; Chen, T.-C.; Wang, J.-C. Regulation of Glucose Homeostasis by Glucocorticoids. In Glucocorticoid Signaling: From Molecules to Mice to Man; Wang, J.-C., Harris, C., Eds.; Springer: New York, NY, USA, 2015; pp. 99–126.
- Facchinello, N.; Skobo, T.; Meneghetti, G.; Colletti, E.; Dinarello, A.; Tiso, N.; Costa, R.; Gioacchini, G.; Carnevali, O.; Argenton, F.; et al. nr3c1 null mutant zebrafish are viable and reveal DNA-binding-independent activities of the glucocorticoid receptor. Sci. Rep. 2017, 7, 4371.
- Hench, P.S.; Kendall, E.C.; Slocumb, C.H.; Polley, H.F. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone: Compound E) and of pituitary Adrenocortical Hormone in Arthritis: Preliminary Report. Ann. Rheum. Dis. 1949, 8, 97–104.
- Perretti, M.; Ahluwalia, A. The microcirculation and inflammation: Site of action for glucocorticoids. Microcirculation 2000, 7, 147–161.
- Smoak, K.A.; Cidlowski, J.A. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech. Ageing Dev. 2004, 125, 697–706.
- Newton, R.; Holden, N. Separating Transrepression and Transactivation: A Distressing Divorce for the Glucocorticoid Receptor? Mol. Pharmacol. 2007, 72, 799–809.
- Necela, B.M.; Cidlowski, J.A. Crystallization of the human glucocorticoid receptor ligand binding domain: A step towards selective glucocorticoids. Trends Pharmacol. Sci. 2003, 24, 58–61.
- Rhen, T.; Cidlowski, J. Antiinflammatory Action of Glucocorticoids—New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723.
- Yeager, M.P.; Guyre, P.M.; Munck, A.U. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol. Scand. 2004, 48, 799–813.
- Kleiman, A.; Tuckermann, J.P. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: Lessons from conditional knockout mice. Mol. Cell. Endocrinol. 2007, 275, 98–108.
- Newton, R.; Shah, S.; Altonsy, M.O.; Gerber, A.N. Glucocorticoid and cytokine crosstalk: Feedback, feedforward, and co-regulatory interactions determine repression or resistance. J. Biol. Chem. 2017, 292, 7163–7172.
- De Bosscher, K. Selective Glucocorticoid Receptor modulators. J. Steroid Biochem. Mol. Biol. 2010, 120, 96–104.
- Chatzopoulou, A.; Schoonheim, P.J.; Torraca, V.; Meijer, A.H.; Spaink, H.; Schaaf, M.J. Functional analysis reveals no transcriptional role for the glucocorticoid receptor β-isoform in zebrafish. Mol. Cell. Endocrinol. 2017, 447, 61–70.
- Schaaf, M.J.M.; Champagne, D.; van Laanen, I.H.C.; van Wijk, D.C.W.A.; Meijer, A.H.; Meijer, O.C.; Spaink, H.P.; Richardson, M.K. Discovery of a Functional Glucocorticoid Receptor β-Isoform in Zebrafish. Endocrinology 2007, 149, 1591–1599.
- Jones, M.T.; Hillhouse, E.W.; Burden, J.L. Dynamics and mechanics of corticosteroid feedback at the hypothalamus and anterior pituitary gland. J. Endocrinol. 1977, 73, 405–417.
- Dallman, M.F.; Akana, S.F.; Jacobson, L.; Levin, N.; Cascio, C.S.; Shinsako, J. Characterization of Corticosterone Feedback Regulation of ACTH Secretion. Ann. N. Y. Acad. Sci. 1987, 512, 402–414.
- Alsop, D.; Vijayan, M.M. Development of the corticosteroid stress axis and receptor expression in zebrafish. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R711–R719.
- Yamamoto, K.R. Steroid receptor regulated transcription of specific genes and gene networks. Annu. Rev. Genet. 1985, 19, 209–252.
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641.
- Miesfeld, R.; Rusconi, S.; Godowski, P.J.; Maler, B.A.; Okret, S.; Wikström, A.-C.; Gustafsson, J.; Yamamoto, K.R. Genetic complementation of a glucocorticoid receptor deficiency by expression of cloned receptor cDNA. Cell 1986, 46, 389–399.
- Jonat, C.; Rahmsdorf, H.J.; Park, K.-K.; Cato, A.; Gebel, S.; Ponta, H.; Herrlich, P. Antitumor promotion and antiinflammation: Down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 1990, 62, 1189–1204.
- Heck, S.; Kullmann, M.; Gast, A.; Ponta, H.; Rahmsdorf, H.; Herrlich, P.; Cato, A. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994, 13, 4087–4095.
- Heck, S.; Bender, K.; Kullmann, M.; Göttlicher, M.; Herrlich, P.; Cato, A.C. Ikappa Balpha -independent downregulation of NF-kappa B activity by glucocorticoid receptor. EMBO J. 1997, 16, 4698–4707.
- Yang-Yen, H.-F.; Chambard, J.C.; Sun, Y.-L.; Smeal, T.; Schmidt, T.; Drouin, J.; Karin, M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62, 1205–1215.
- Nikolaus, S.; Fölscn, U.; Schreiber, S. Immunopharmacology of 5-aminosalicylic acid and of glucocorticoids in the therapy of inflammatory bowel disease. Hepatogastroenterology 2000, 47, 71–82.
- Neeck, G.; Renkawitz, R.; Eggert, M. Molecular aspects of glucocorticoid hormone action in rheumatoid arthritis. Cytokines Cell. Mol. Ther. 2002, 7, 61–69.
- Chrousos, G.P.; Kino, T. Intracellular Glucocorticoid Signaling: A Formerly Simple System Turns Stochastic. Sci. STKE 2005, 2005, pe48.
- Revollo, J.R.; Cidlowski, J.A. Mechanisms Generating Diversity in Glucocorticoid Receptor Signaling. Ann. N. Y. Acad. Sci. 2009, 1179, 167–178.
- Busillo, J.M.; Cidlowski, J.A. The five Rs of glucocorticoid action during inflammation: Ready, reinforce, repress, resolve, and restore. Trends Endocrinol. Metab. 2013, 24, 109–119.
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Lühder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921.
- Van Staa, T.P.; Leufkens, H.G.M.; Abenhaim, L.; Begaud, B.; Zhang, B.; Cooper, C. Use of oral corticosteroids in the United Kingdom. QJM Int. J. Med. 2000, 93, 105–111.
- Moghadam-Kia, S.; Werth, V.P. Prevention and treatment of systemic glucocorticoid side effects. Int. J. Dermatol. 2010, 49, 239–248.
- Barnes, P.J.; Adcock, I. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917.
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2010, 163, 29–43.
- Schaaf, M.J.; Cidlowski, J.A. Molecular mechanisms of glucocorticoid action and resistance. J. Steroid Biochem. Mol. Biol. 2002, 83, 37–48.
- Spies, L.-M.L.; Verhoog, N.J.D.; Louw, A. Acquired Glucocorticoid Resistance Due to Homologous Glucocorticoid Receptor Downregulation: A Modern Look at an Age-Old Problem. Cells 2021, 10, 2529.
- Howell, M.P.; Muglia, L.J. Effects of genetically altered brain glucocorticoid receptor action on behavior and adrenal axis regulation in mice. Front. Neuroendocrinol. 2006, 27, 275–284.
- Zappia, C.D.; Torralba-Agu, V.; Echeverria, E.; Fitzsimons, C.P.; Fernández, N.; Monczor, F. Antihistamines Potentiate Dexamethasone Anti-Inflammatory Effects. Impact on Glucocorticoid Receptor-Mediated Expression of Inflammation-Related Genes. Cells 2021, 10, 3026.
- Bamberger, C.M.; Schulte, H.M.; Chrousos, G.P. Molecular Determinants of Glucocorticoid Receptor Function and Tissue Sensitivity to Glucocorticoids. Endocr. Rev. 1996, 17, 245–261.
- Faught, E.; Vijayan, M.M. Maternal stress and fish reproduction: The role of cortisol revisited. Fish Fish. 2018, 19, 1016–1030.
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The Multifaceted Mineralocorticoid Receptor. Compr. Physiol. 2014, 4, 965–994.
- Spach, C.; Streeten, D.H.P. Retardation of Sodium Exchange in Dog Erythrocytes by Physiological Concentrations of Aldosterone, in Vitro. J. Clin. Investig. 1964, 43, 217–227.
- Moura, A.M.; Worcel, M. Direct action of aldosterone on transmembrane 22Na efflux from arterial smooth muscle. Rapid and delayed effects. Hypertension 1984, 6, 425–430.
- Buttgereit, F.; Wehling, M.; Burmester, G.R. A new hypothesis of modular glucocorticoid actions: Steroid treatment of rheumatic diseases revisited. Arthritis Rheum. 1998, 41, 761–767.
- Buttgereit, F.; Scheffold, A. Rapid glucocorticoid effects on immune cells. Steroids 2002, 67, 529–534.
- Pereda, M.P.; Lohrer, P.; Kovalovsky, D.; Castro, C.P.; Goldberg, V.; Losa, M.; Chervin, A.; Berner, S.; Molina, H.; Stalla, G.K.; et al. Interleukin-6 is inhibited by glucocorticoids and stimulates ACTH secretion and POMC expression in human corticotroph pituitary adenomas. Exp. Clin. Endocrinol. Diabetes 2000, 108, 202–207.
- Cruz-Topete, D.; Cidlowski, J. One Hormone, Two Actions: Anti- and Pro-Inflammatory Effects of Glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32.
- Arnett, M.G.; Muglia, L.M.; Laryea, G.; Muglia, L.J. Genetic Approaches to Hypothalamic-Pituitary-Adrenal Axis Regulation. Neuropsychopharmacology 2015, 41, 245–260.
- Bertini, R.; Bianchi, M.; Ghezzi, P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J. Exp. Med. 1988, 167, 1708–1712.
- Edwards, C.K., 3rd; Yunger, L.M.; Lorence, R.M.; Dantzer, R.; Kelley, K.W. The pituitary gland is required for protection against lethal effects of Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 1991, 88, 2274–2277.
- Macphee, I.A.M.; Antoni, F.A.; Mason, D.W. Spontaneous recovery of rats from experimental allergic encephalomyelitis is dependent on regulation of the immune system by endogenous adrenal corticosteroids. J. Exp. Med. 1989, 169, 431–445.
- Ramachandra, R.N. Neuro-hormonal host defence in endotoxin shock. Brain Behav. Immun. 1992, 6, 157–169.
- Refojo, D.; Liberman, A.C.; Giacomini, D.; Nagashima, A.C.; Graciarena, M.; Echenique, C.; Pereda, M.P.; Stalla, G.; Holsboer, F.; Arzt, E. Integrating Systemic Information at the Molecular Level: Cross-talk between steroid receptors and cytokine signaling on different target cells. Ann. N. Y. Acad. Sci. 2003, 992, 196–204.
- Ruzek, M.C.; Pearce, B.D.; Miller, A.H.; Biron, C.A. Endogenous glucocorticoids protect against cytokine-mediated lethality during viral infection. J. Immunol. 1999, 162, 3527–3533.
- Silverman, M.N.; Sternberg, E.M. Glucocorticoid regulation of inflammation and its functional correlates: From HPA axis to glucocorticoid receptor dysfunction. Ann. N. Y. Acad. Sci. 2012, 1261, 55–63.
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialog. Clin. Neurosci. 2006, 8, 383–395.
- De Guia, R.; Rose, A.J.; Herzig, S. Glucocorticoid hormones and energy homeostasis. Horm. Mol. Biol. Clin. Investig. 2014, 19, 117–128.
- Katsu, Y.; Iguchi, T. Subchapter 95D—Cortisol. In Handbook of Hormones; Takei, Y., Ando, H., Tsutsui, K., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 532–533.
- Cummins, E.P.; Taylor, C.T. Hypoxia-responsive transcription factors. Pflügers Arch. 2005, 450, 363–371.
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975.
- Dougherty, E.J.; Pollenz, R.S. ARNT: A Key bHLH/PAS Regulatory Protein Across Multiple Pathways. Compr. Toxicol. 2010, 2, 231–252.
- Pelster, B.; Egg, M. Hypoxia-inducible transcription factors in fish: Expression, function and interconnection with the circadian clock. J. Exp. Biol. 2018, 221, jeb163709.
- Hill, A.J.; Heiden, T.C.K.; Heideman, W.; Peterson, R.E. Potential Roles of Arnt2 in Zebrafish Larval Development. Zebrafish 2009, 6, 79–91.
- Prasch, A.L.; Tanguay, R.L.; Mehta, V.; Heideman, W.; Peterson, R.E. Identification of Zebrafish ARNT1 Homologs: 2,3,7,8-Tetrachlorodibenzo-p-dioxin Toxicity in the Developing Zebrafish Requires ARNT1. Mol. Pharmacol. 2005, 69, 776–787.
- Elks, P.M.; Renshaw, S.; Meijer, A.H.; Walmsley, S.; van Eeden, F.J. Exploring the HIFs, buts and maybes of hypoxia signalling in disease: Lessons from zebrafish models. Dis. Model. Mech. 2015, 8, 1349–1360.
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408.
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471.
- van Rooijen, E.; Voest, E.E.; Logister, I.; Korving, J.; Schwerte, T.; Schulte-Merker, S.; Giles, R.H.; van Eeden, F.J. Zebrafish mutants in the von Hippel-Lindau tumor suppressor display a hypoxic response and recapitulate key aspects of Chuvash polycythemia. Blood 2009, 113, 6449–6460.
- Pescador, N.; Cuevas, Y.; Naranjo, S.; Alcaide, M.; Villar, D.; Landázuri, M.O.; del Peso, L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005, 390, 189–197.
- Santhakumar, K.; Judson, E.C.; Elks, P.M.; McKee, S.; Elworthy, S.; Van Rooijen, E.; Walmsley, S.S.; Renshaw, S.A.; Cross, S.S.; Van Eeden, F.J. A Zebrafish Model to Study and Therapeutically Manipulate Hypoxia Signaling in Tumorigenesis. Cancer Res. 2012, 72, 4017–4027.
- Basu, M.; Sawhney, R.C.; Kumar, S.; Pal, K.; Prasad, R.; Selvamurthy, W. Glucocorticoids as prophylaxis against acute mountain sickness. Clin. Endocrinol. 2002, 57, 761–767.
- Leonard, M.O.; Godson, C.; Brady, H.R.; Taylor, C. Potentiation of Glucocorticoid Activity in Hypoxia through Induction of the Glucocorticoid Receptor. J. Immunol. 2005, 174, 2250–2257.
- Kodama, T.; Shimizu, N.; Yoshikawa, N.; Makino, Y.; Ouchida, R.; Okamoto, K.; Hisada, T.; Nakamura, H.; Morimoto, C.; Tanaka, H. Role of the Glucocorticoid Receptor for Regulation of Hypoxia-dependent Gene Expression. J. Biol. Chem. 2003, 278, 33384–33391.
- Wagner, A.; Huck, G.; Stiehl, D.; Jelkmann, W.; Hellwig-Bürgel, T. Dexamethasone impairs hypoxia-inducible factor-1 function. Biochem. Biophys. Res. Commun. 2008, 372, 336–340.
- Sun, Y.-Y.; Wang, C.-Y.; Hsu, M.-F.; Juan, S.-H.; Chang, C.-Y.; Chou, C.-M.; Yang, L.-Y.; Hung, K.-S.; Xu, J.; Lee, Y.-H.; et al. Glucocorticoid Protection of Oligodendrocytes against Excitotoxin Involving Hypoxia-Inducible Factor-1 in a Cell-Type-Specific Manner. J. Neurosci. 2010, 30, 9621–9630.
- Elks, P.; Van Eeden, F.J.; Dixon, G.; Wang, X.; Reyes-Aldasoro, C.C.; Ingham, P.W.; Whyte, M.K.B.; Walmsley, S.; Renshaw, S.A. Activation of hypoxia-inducible factor-1α (Hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood 2011, 118, 712–722.
- Nesan, D.; Vijayan, M.M. Maternal Cortisol Mediates Hypothalamus-Pituitary-Interrenal Axis Development in Zebrafish. Sci. Rep. 2016, 6, 22582.
- Tsigos, C.; Chrousos, G.P. Hypothalamic–pituitary–adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871.
- Schmidt, F.; Braunbeck, T. Alterations along the Hypothalamic-Pituitary-Thyroid Axis of the Zebrafish (Danio rerio) after Exposure to Propylthiouracil. J. Thyroid. Res. 2011, 2011, 376243.
- Gonzalez-Nunez, V. Identification of two proopiomelanocortin genes in zebrafish (Danio rerio). Mol. Brain Res. 2003, 120, 1–8.
- Hansen, I.A.; To, T.T.; Wortmann, S.; Burmester, T.; Winkler, C.; Meyer, S.R.; Neuner, C.; Fassnacht, M.; Allolio, B. The pro-opiomelanocortin gene of the zebrafish (Danio rerio). Biochem. Biophys. Res. Commun. 2003, 303, 1121–1128.
- To, T.T.; Hahner, S.; Nica, G.; Rohr, K.B.; Hammerschmidt, M.; Winkler, C.; Allolio, B. Pituitary-Interrenal Interaction in Zebrafish Interrenal Organ Development. Mol. Endocrinol. 2007, 21, 472–485.
- Wagle, M.; Mathur, P.; Guo, S. Corticotropin-Releasing Factor Critical for Zebrafish Camouflage Behavior Is Regulated by Light and Sensitive to Ethanol. J. Neurosci. 2011, 31, 214–224.
- Shi, C.; Lu, Y.; Zhai, G.; Huang, J.; Shang, G.; Lou, Q.; Li, D.; Jin, X.; He, J.; Du, Z.; et al. Hyperandrogenism in POMCa-deficient zebrafish enhances somatic growth without increasing adiposity. J. Mol. Cell Biol. 2019, 12, 291–304.
- Mommsen, T.P.; Vijayan, M.M.; Moon, T.W. Cortisol in teleosts: Dynamics, mechanisms of action, and metabolic regulation. Rev. Fish Biol. Fish. 1999, 9, 211–268.
- Nesan, D.; Vijayan, M.M. Role of glucocorticoid in developmental programming: Evidence from zebrafish. Gen. Comp. Endocrinol. 2013, 181, 35–44.
- Spiga, F.; Walker, J.J.; Terry, J.R.; Lightman, S.L. HPA Axis-Rhythms. Compr. Physiol. 2014, 4, 1273–1298.
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids. Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31.
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416.
- Dallman, M.F.; Akana, S.F.; Cascio, C.S.; Darlington, D.N.; Jacobson, L.; Levin, N. Regulation of ACTH Secretion: Variations on a Theme of B. Recent Prog. Horm. Res. 1987, 43, 113–173.
- Drouin, J.; Trifiro, M.A.; Plante, R.K.; Nemer, M.; Eriksson, P.; Wrange, O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene transcription. Mol. Cell. Biol. 1989, 9, 5305–5314.
- Ulrich-Lai, Y.; Herman, J. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 2009, 10, 397–409.
- Nicolaides, N.C.; Galata, Z.; Kino, T.; Chrousos, G.P.; Charmandari, E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010, 75, 1–12.
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the Stress System and the Role of Glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19.
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839.
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475.
- Schoneveld, O.J.; Gaemers, I.C.; Lamers, W.H. Mechanisms of glucocorticoid signalling. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2004, 1680, 114–128.
- Oakley, R.H.; Cidlowski, J.A. The Biology of the Glucocorticoid Receptor: New Signaling Mechanism in Health and Disease. J. Allergy Clin. Inmunol. 2013, 132, 1033–1044.
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895.
- Kumar, R.; Thompson, E.B. Gene regulation by the glucocorticoid receptor: Structure:function relationship. J. Steroid Biochem. Mol. Biol. 2005, 94, 383–394.
- Glass, C.K.; Rose, D.W.; Rosenfeld, M.G. Nuclear receptor coactivators. Curr. Opin. Cell Biol. 1997, 9, 222–232.
- Beck, I.; Berghe, W.V.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in Inflammation: The Interplay of Glucocorticoid Receptor-Based Mechanisms and Kinases and Phosphatases. Endocr. Rev. 2009, 30, 830–882.
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M.; et al. Crystal Structure of the Glucocorticoid Receptor Ligand Binding Domain Reveals a Novel Mode of Receptor Dimerization and Coactivator Recognition. Cell 2002, 110, 93–105.
- Yudt, M.R.; Cidlowski, J.A. Molecular Identification and Characterization of A and B Forms of the Glucocorticoid Receptor. Mol. Endocrinol. 2001, 15, 1093–1103.
- Yudt, M.R.; Cidlowski, J. The Glucocorticoid Receptor: Coding a Diversity of Proteins and Responses through a Single Gene. Mol. Endocrinol. 2002, 16, 1719–1726.
- Oakley, R.H.; Cidlowski, J.A. Cellular Processing of the Glucocorticoid Receptor Gene and Protein: New Mechanisms for Generating Tissue-specific Actions of Glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184.
- Merkulov, V.M.; Merkulova, T.I. Glucocorticoid receptor isoforms generated by alternative splicing and alternative translation initiation. Russ. J. Genet. Appl. Res. 2012, 2, 205–213.
- Rafacho, A.; Ortsäter, H.; Nadal, A.; Quesada, I. Glucocorticoid treatment and endocrine pancreas function: Implications for glucose homeostasis, insulin resistance and diabetes. J. Endocrinol. 2014, 223, R49–R62.
- Marchi, D. Bidirectional Crosstalk between Hypoxia-Inducible Factor and Glucocorticoid Signalling in Zebrafish Larvae. University of Sheffield. 2020. Available online: https://etheses.whiterose.ac.uk/28224/ (accessed on 2 December 2021).
- Encío, I.J.; Detera-Wadleigh, S.D. The genomic structure of the human glucocorticoid receptor. J. Biol. Chem. 1991, 266, 7182–7188.
- Yudt, M.R.; Jewell, C.M.; Bienstock, R.; Cidlowski, J.A. Molecular Origins for the Dominant Negative Function of Human Glucocorticoid Receptor Beta. Mol. Cell. Biol. 2003, 23, 4319–4330.
- Oakley, R.H.; Sar, M.; Cidlowski, J.A. The Human Glucocorticoid Receptor β Isoform: Expression, biochemical properties, and putative function. J. Biol. Chem. 1996, 271, 9550–9559.
- Bamberger, C.M.; Bamberger, A.M.; de Castro, M.; Chrousos, G.P. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J. Clin. Investig. 1995, 95, 2435–2441.
- Oakley, R.H.; Jewell, C.M.; Yudt, M.R.; Bofetiado, D.M.; Cidlowski, J. The Dominant Negative Activity of the Human Glucocorticoid Receptor β Isoform. Specificity and mechanisms of action. J. Biol. Chem. 1999, 274, 27857–27866.
- Charmandari, E.; Chrousos, G.P.; Ichijo, T.; Bhattacharyya, N.; Vottero, A.; Souvatzoglou, E.; Kino, T. The Human Glucocorticoid Receptor (hGR) β Isoform Suppresses the Transcriptional Activity of hGRα by Interfering with Formation of Active Coactivator Complexes. Mol. Endocrinol. 2005, 19, 52–64.
- Hauk, P.J.; Goleva, E.; Strickland, I.; Vottero, A.; Chrousos, G.P.; Kisich, K.O.; Leung, D.Y.M. Increased Glucocorticoid ReceptorβExpression Converts Mouse Hybridoma Cells to a Corticosteroid-Insensitive Phenotype. Am. J. Respir. Cell Mol. Biol. 2002, 27, 361–367.
- Strickland, I.; Kisich, K.; Hauk, P.J.; Vottero, A.; Chrousos, G.P.; Klemm, D.J.; Leung, D.Y. High Constitutive Glucocorticoid Receptor β in Human Neutrophils Enables Them to Reduce Their Spontaneous Rate of Cell Death in Response to Corticosteroids. J. Exp. Med. 2001, 193, 585–594.
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545.
- Lewis-Tuffin, L.J.; Jewell, C.M.; Bienstock, R.; Collins, J.B.; Cidlowski, J. Human Glucocorticoid Receptor β Binds RU-486 and Is TranscriptionallyActive. Mol. Cell. Biol. 2007, 27, 2266–2282.
- Kino, T.; Manoli, I.; Kelkar, S.; Wang, Y.; Su, Y.A.; Chrousos, G.P. Glucocorticoid receptor (GR) β has intrinsic, GRα-independent transcriptional activity. Biochem. Biophys. Res. Commun. 2009, 381, 671–675.
- Honda, M.; Orii, F.; Ayabe, T.; Imai, S.; Ashida, T.; Obara, T.; Kohgo, Y. Expression of glucocorticoid receptor β in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology 2000, 118, 859–866.
- Shahidi, H.; Vottero, A.; Stratakis, C.A.; Taymans, S.E.; Karl, M.; Longui, C.A.; Chrousos, G.P.; Daughaday, W.H.; Gregory, S.A.; Plate, J.M. Imbalanced Expression of the Glucocorticoid Receptor Isoforms in Cultured Lymphocytes from a Patient with Systemic Glucocorticoid Resistance and Chronic Lymphocytic Leukemia. Biochem. Biophys. Res. Commun. 1999, 254, 559–565.
- Bergeron, C.; Fukakusa, M.; Olivenstein, R.; Lemiere, C.; Shannon, J.; Ernst, P.; Martin, J.G.; Hamid, Q. Increased glucocorticoid receptor–β expression, but not decreased histone deacetylase 2, in severe asthma. J. Allergy Clin. Immunol. 2006, 117, 703–705.
- Hamid, Q.A.; Wenzel, S.E.; Hauk, P.J.; Tsicopoulos, A.; Wallaert, B.; Lafitte, J.-J.; Chrousos, G.P.; Szefler, S.J.; Leung, D.Y.M. Increased Glucocorticoid Receptor β in Airway Cells of Glucocorticoid-insensitive Asthma. Am. J. Respir. Crit. Care Med. 1999, 159, 1600–1604.
- Goleva, E.; Li, L.-B.; Eves, P.T.; Strand, M.; Martin, R.J.; Leung, D.Y.M. Increased Glucocorticoid Receptor β Alters Steroid Response in Glucocorticoid-insensitive Asthma. Am. J. Respir. Crit. Care Med. 2006, 173, 607–616.
- Leung, D.Y.; Hamid, Q.; Vottero, A.; Szefler, S.J.; Surs, W.; Minshall, E.; Chrousos, G.P.; Klemm, D.J. Association of Glucocorticoid Insensitivity with Increased Expression of Glucocorticoid Receptor β. J. Exp. Med. 1997, 186, 1567–1574.
- Chatzopoulou, A.; Roy, U.; Meijer, A.H.; Alia, A.; Spaink, H.; Schaaf, M.J.M. Transcriptional and Metabolic Effects of Glucocorticoid Receptor α and β Signaling in Zebrafish. Endocrinology 2015, 156, 1757–1769.
- Ramos-Ramírez, P.; Tliba, O. Glucocorticoid Receptor β (GRβ): Beyond Its Dominant-Negative Function. Int. J. Mol. Sci. 2021, 22, 3649.
- Schaaf, M.; Chatzopoulou, A.; Spaink, H. The zebrafish as a model system for glucocorticoid receptor research. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2009, 153, 75–82.
- Holownia, A.; Mroz, R.M.; Kolodziejczyk, A.; Chyczewska, E.; Braszko, J.J. Increased FKBP51 in induced sputum cells of chronic obstructive pulmonary disease patients after therapy. Eur. J. Med. Res. 2009, 14 (Suppl. 4), 108–111.
- Storer, C.L.; Dickey, C.A.; Galigniana, M.D.; Rein, T.; Cox, M.B. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol. Metab. 2011, 22, 481–490.
- Woodruff, P.G.; Boushey, H.A.; Dolganov, G.M.; Barker, C.S.; Yang, J.; Donnelly, S.; Ellwanger, A.; Sidhu, S.S.; Dao-Pick, T.P.; Pantoja, C.; et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc. Natl. Acad. Sci. USA 2007, 104, 15858–15863.
- Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009, 34, S186–S195.
- Liberman, A.C.; Budziñski, M.L.; Sokn, C.; Gobbini, R.P.; Steininger, A.; Arzt, E. Regulatory and Mechanistic Actions of Glucocorticoids on T and Inflammatory Cells. Front. Endocrinol. 2018, 9, 235.
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New Insights into the Anti-inflammatory Mechanisms of Glucocorticoids: An Emerging Role for Glucocorticoid-Receptor-Mediated Transactivation. Endocrinology 2013, 154, 993–1007.
- Presman, D.M.; Hager, G.L. More than meets the dimer: What is the quaternary structure of the glucocorticoid receptor? Transcription 2016, 8, 32–39.
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA Binding of the Glucocorticoid Receptor Is Not Essential for Survival. Cell 1998, 93, 531–541.
- Prager, E.M.; Johnson, L.R. Stress at the Synapse: Signal Transduction Mechanisms of Adrenal Steroids at Neuronal Membranes. Sci. Signal. 2009, 2, re5.
- Groeneweg, F.L.; Karst, H.; de Kloet, R.; Joels, M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol. 2011, 209, 153–167.
- Uhlenhaut, H.; Barish, G.D.; Yu, R.T.; Downes, M.; Karunasiri, M.; Liddle, C.; Schwalie, P.C.; Hübner, N.; Evans, R.M. Insights into Negative Regulation by the Glucocorticoid Receptor from Genome-wide Profiling of Inflammatory Cistromes. Mol. Cell 2012, 49, 158–171.
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101.
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread Negative Response Elements Mediate Direct Repression by Agonist- Liganded Glucocorticoid Receptor. Cell 2011, 145, 224–241.
- Ramamoorthy, S.; Cidlowski, J.A. Exploring the Molecular Mechanisms of Glucocorticoid Receptor Action from Sensitivity to Resistance. Endocr. Dev. 2013, 24, 41–56.
- Escoter-Torres, L.; Caratti, G.; Mechtidou, A.; Tuckermann, J.; Uhlenhaut, N.H.; Vettorazzi, S. Fighting the Fire: Mechanisms of Inflammatory Gene Regulation by the Glucocorticoid Receptor. Front. Immunol. 2019, 10, 1859.
- Oh, K.-S.; Patel, H.; Gottschalk, R.A.; Lee, W.S.; Baek, S.; Fraser, I.; Hager, G.L.; Sung, M.-H. Anti-Inflammatory Chromatinscape Suggests Alternative Mechanisms of Glucocorticoid Receptor Action. Immunity 2017, 47, 298–309.e5.
- Ratman, D.; Berghe, W.V.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.; De Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54.
- Baudrand, R.; Pojoga, L.; Romero, J.R. Chapter 10—Aldosterone’s Mechanism of Action: Genomic and Nongenomic Signaling. In Textbook of Nephro-Endocrinology, 2nd ed.; Singh, A.K., Williams, G.H., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 173–188.
- Reul, J.M.H.M.; de Kloet, R. Two Receptor Systems for Corticosterone in Rat Brain: Microdistribution and Differential Occupation. Endocrinology 1985, 117, 2505–2511.
- Reul, J.M.; Collins, A.; Saliba, R.S.; Mifsud, K.; Carter, S.D.; Gutierrez-Mecinas, M.; Qian, X.; Linthorst, A.C. Glucocorticoids, epigenetic control and stress resilience. Neurobiol. Stress 2014, 1, 44–59.
- Grossmann, C.; Gekle, M. Non-classical actions of the mineralocorticoid receptor: Misuse of EGF receptors? Mol. Cell. Endocrinol. 2007, 277, 6–12.
- Sireeni, J.; Bakker, N.; Jaikumar, G.; Obdam, D.; Slabbekoorn, H.; Tudorache, C.; Schaaf, M. Profound effects of glucocorticoid resistance on anxiety-related behavior in zebrafish adults but not in larvae. Gen. Comp. Endocrinol. 2020, 292, 113461.
- Müller, M.B.; Holsboer, F.; Keck, M.E. Genetic modification of corticosteroid receptor signalling: Novel insights into pathophysiology and treatment strategies of human affective disorders. Neuropeptides 2002, 36, 117–131.
- Van Eekelen, J.; Bohn, M.; De Kloet, E. Postnatal ontogeny of mineralocorticoid and glucocorticoid receptor gene expression in regions of the rat tel- and diencephalon. Dev. Brain Res. 1991, 61, 33–43.
- Hartmann, J.; Bajaj, T.; Klengel, C.; Chatzinakos, C.; Ebert, T.; Dedic, N.; McCullough, K.M.; Lardenoije, R.; Joëls, M.; Meijer, O.C.; et al. Mineralocorticoid receptors dampen glucocorticoid receptor sensitivity to stress via regulation of FKBP5. Cell Rep. 2021, 35, 109185.
- De Kloet, E.; Meijer, O.; de Nicola, A.; de Rijk, R.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 2018, 49, 124–145.
- Lee, H.B.; Schwab, T.L.; Sigafoos, A.N.; Gauerke, J.L.; Ii, R.G.K.; Serres, M.R.; Jacobs, D.C.; Cotter, R.P.; Das, B.; Petersen, M.O.; et al. Novel zebrafish behavioral assay to identify modifiers of the rapid, nongenomic stress response. Genes Brain Behav. 2018, 18, e12549.
- Marchi, D.; Santhakumar, K.; Markham, E.; Li, N.; Storbeck, K.-H.; Krone, N.; Cunliffe, V.T.; van Eeden, F.J.M. Bidirectional crosstalk between Hypoxia-Inducible Factor and glucocorticoid signalling in zebrafish larvae. PLoS Genet. 2020, 16, e1008757.
- Ince, L.M.; Weber, J.; Scheiermann, C. Control of Leukocyte Trafficking by Stress-Associated Hormones. Front. Immunol. 2019, 9, 3143.
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13.
- Schmidt, S.; Rainer, J.; Ploner, C.; Presul, E.; Riml, S.; Kofler, R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: Molecular mechanisms and clinical relevance. Cell Death Differ. 2004, 11, S45–S55.
- Planey, S.L.; Litwack, G. Glucocorticoid-Induced Apoptosis in Lymphocytes. Biochem. Biophys. Res. Commun. 2000, 279, 307–312.
- Ashwell, J.D.; Lu, F.W.M.; Vacchio, M.S. Glucocorticoids in T Cell Development and Function. Annu. Rev. Immunol. 2000, 18, 309–345.
- Herold, M.J.; McPherson, K.G.; Reichardt, H.M. Glucocorticoids in T cell apoptosis and function. Cell. Mol. Life Sci. 2005, 63, 60–72.
- McColl, A.; Bournazos, S.; Franz, S.; Perretti, M.; Morgan, B.P.; Haslett, C.; Dransfield, I. Glucocorticoids Induce Protein S-Dependent Phagocytosis of Apoptotic Neutrophils by Human Macrophages. J. Immunol. 2009, 183, 2167–2175.
- Miller, A.H.; Spencer, R.L.; Stein, M.; McEwen, B.S. Adrenal steroid receptor binding in spleen and thymus after stress or dexamethasone. Am. J. Physiol. Metab. 1990, 259, E405.
- Barish, G.D.; Downes, M.; Alaynick, W.A.; Yu, R.T.; Ocampo, C.B.; Bookout, A.L.; Mangelsdorf, D.J.; Evans, R.M. A Nuclear Receptor Atlas: Macrophage Activation. Mol. Endocrinol. 2005, 19, 2466–2477.
- Lim, H.-Y.; Müller, N.; Herold, M.J.; Brandt, J.V.D.; Reichardt, H.M. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 2007, 122, 47–53.
- Harizi, H.; Mormede, P.; Corcuff, J. Inter-strain differences in glucocorticoid and mineralocorticoid effects on macrophage and lymphocyte functions in mice. J. Neuroimmunol. 2008, 204, 38–42.
- Bene, N.C.; Alcaide, P.; Wortis, H.H.; Jaffe, I.Z. Mineralocorticoid receptors in immune cells: Emerging role in cardiovascular disease. Steroids 2014, 91, 38–45.
- Muñoz-Durango, N.; Vecchiola, A.; Gonzalez-Gomez, L.M.; Simon, F.; Riedel, C.; Fardella, C.E.; Kalergis, A.M. Modulation of Immunity and Inflammation by the Mineralocorticoid Receptor and Aldosterone. BioMed Res. Int. 2015, 2015, 652738.
- Van Der Heijden, C.D.C.C.; Deinum, J.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc. Res. 2018, 114, 944–953.
- Vandevyver, S.; Dejager, L.; Libert, C. On the Trail of the Glucocorticoid Receptor: Into the Nucleus and Back. Traffic 2011, 13, 364–374.
- Ronchetti, S.; Migliorati, G.; Riccardi, C. GILZ as a Mediator of the Anti-Inflammatory Effects of Glucocorticoids. Front. Endocrinol. 2015, 6, 170.
- Scheinman, R.I.; Gualberto, A.; Jewell, C.M.; Cidlowski, J.A.; Baldwin, A.S. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol. Cell. Biol. 1995, 15, 943–953.
- Escoter-Torres, L.; Greulich, F.; Quagliarini, F.; Wierer, M.; Uhlenhaut, N.H. Anti-inflammatory functions of the glucocorticoid receptor require DNA binding. Nucleic Acids Res. 2020, 48, 8393–8407.
- Stellato, C. Post-transcriptional and Nongenomic Effects of Glucocorticoids. Proc. Am. Thorac. Soc. 2004, 1, 255–263.
- De Bosscher, K.; Van Craenenbroeck, K.; Meijer, O.C.; Haegeman, G. Selective transrepression versus transactivation mechanisms by glucocorticoid receptor modulators in stress and immune systems. Eur. J. Pharmacol. 2008, 583, 290–302.
- De Bosscher, K.; Haegeman, G.; Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 2010, 10, 497–504.
- Davies, T.H.; Ning, Y.-M.; Sánchez, E.R. Differential Control of Glucocorticoid Receptor Hormone-Binding Function by Tetratricopeptide Repeat (TPR) Proteins and the Immunosuppressive Ligand FK506. Biochemistry 2005, 44, 2030–2038.
- Liberman, A.C.; Druker, J.; Perone, M.J.; Arzt, E. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine Growth Factor Rev. 2007, 18, 45–56.
- Pascual, G.; Glass, C.K. Nuclear receptors versus inflammation: Mechanisms of transrepression. Trends Endocrinol. Metab. 2006, 17, 321–327.
- Limbourg, F.P.; Liao, J.K. Nontranscriptional actions of the glucocorticoid receptor. J. Mol. Med. 2003, 81, 168–174.
- Liberman, A.C.; Refojo, D.; Druker, J.; Toscano, M.; Rein, T.; Holsboer, F.; Arzt, E. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J. 2007, 21, 1177–1188.
- Vayssière, B.M.; Dupont, S.; Choquart, A.; Petit, F.; Garcia, T.; Marchandeau, C.; Gronemeyer, H.; Resche-Rigon, M. Synthetic Glucocorticoids That Dissociate Transactivation and AP-1 Transrepression Exhibit Antiinflammatory Activity in Vivo. Mol. Endocrinol. 1997, 11, 1245–1255.
- Belvisi, M.G.; Wicks, S.L.; Battram, C.H.; Bottoms, S.E.W.; Redford, J.E.; Woodman, P.; Brown, T.J.; Webber, S.E.; Foster, M.L. Therapeutic Benefit of a Dissociated Glucocorticoid and the Relevance of In Vitro Separation of Transrepression from Transactivation Activity. J. Immunol. 2001, 166, 1975–1982.
- Schacke, H.; Schottelius, A.; Docke, W.-D.; Strehlke, P.; Jaroch, S.; Schmees, N.; Rehwinkel, H.; Hennekes, H.; Asadullah, K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc. Natl. Acad. Sci. USA 2003, 101, 227–232.
- De Bosscher, K.; Beck, I.M.; Dejager, L.; Bougarne, N.; Gaigneaux, A.; Chateauvieux, S.; Ratman, D.; Bracke, M.; Tavernier, J.; Berghe, W.V.; et al. Selective modulation of the glucocorticoid receptor can distinguish between transrepression of NF-κB and AP-1. Cell. Mol. Life Sci. 2013, 71, 143–163.
- De Bosscher, K.; Berghe, W.V.; Beck, I.; Van Molle, W.; Hennuyer, N.; Hapgood, J.; Libert, C.; Staels, B.; Louw, A.; Haegeman, G. A fully dissociated compound of plant origin for inflammatory gene repression. Proc. Natl. Acad. Sci. USA 2005, 102, 15827–15832.
- Duque, E.D.A.; Munhoz, C.D. The Pro-inflammatory Effects of Glucocorticoids in the Brain. Front. Endocrinol. 2016, 7, 78.
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. Regulatory Network Analyses Reveal Genome-Wide Potentiation of LIF Signaling by Glucocorticoids and Define an Innate Cell Defense Response. PLoS Genet. 2008, 4, e1000224.
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. The Stat3/GR Interaction Code: Predictive Value of Direct/Indirect DNA Recruitment for Transcription Outcome. Mol. Cell 2012, 47, 38–49.
- Dittrich, A.; Khouri, C.; Sackett, S.D.; Ehlting, C.; Böhmer, O.; Albrecht, U.; Bode, J.G.; Trautwein, C.; Schaper, F. Glucocorticoids increase interleukin-6-dependent gene induction by interfering with the expression of the suppressor of cytokine signaling 3 feedback inhibitor. Hepatology 2011, 55, 256–266.
- Xie, Y.; Tolmeijer, S.; Oskam, J.M.; Tonkens, T.; Meijer, A.H.; Schaaf, M.J.M. Glucocorticoids inhibit macrophage differentiation towards a pro-inflammatory phenotype upon wounding without affecting their migration. Dis. Model. Mech. 2019, 12.
- Bos, R.V.D.; Cromwijk, S.; Tschigg, K.; Althuizen, J.; Zethof, J.; Whelan, R.; Flik, G.; Schaaf, M. Early Life Glucocorticoid Exposure Modulates Immune Function in Zebrafish (Danio rerio) Larvae. Front. Immunol. 2020, 11, 727.
- John, S.; Sabo, P.J.; Thurman, R.E.; Sung, M.-H.; Biddie, S.; Johnson, T.A.; Hager, G.L.; Stamatoyannopoulos, J.A. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011, 43, 264–268.
- John, S.; Sabo, P.J.; Johnson, T.A.; Sung, M.-H.; Biddie, S.C.; Lightman, S.L.; Voss, T.C.; Davis, S.R.; Meltzer, P.S.; Stamatoyannopoulos, J.A.; et al. Interaction of the Glucocorticoid Receptor with the Chromatin Landscape. Mol. Cell 2008, 29, 611–624.
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid Receptor Recruitment of Histone Deacetylase 2 Inhibits Interleukin-1β-Induced Histone H4 Acetylation on Lysines 8 and 12. Mol. Cell. Biol. 2000, 20, 6891–6903.
More
Information
Subjects:
Immunology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
856
Revisions:
3 times
(View History)
Update Date:
23 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No