Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Davide Marchi and Version 3 by Camila Xu.
Glucocorticoids (GCs) represent a well-known class of lipophilic steroid hormones biosynthesised, with a circadian rhythm, by the adrenal glands in humans and by the inter-renal tissue in teleost fish (e.g., zebrafish).
- glucocorticoid
- glucocorticoid receptor
- hypoxia inducible factor
- crosstalk
- inflammation
- immune modulations
1. Introduction
The name “glucocorticoid” (GC) is a portmanteau word (glucose + cortex + steroid), which derives from their key role in the regulation of glucose metabolism, their biosynthesis at the level of the adrenal cortex and their steroidal structure. They represent a well-known class of lipophilic steroid hormones synthetized, with a circadian rhythm, by the adrenal glands in humans and by the inter-renal tissue in teleost fish. GC circadian production in mammals is tuned by the hypothalamus-pituitary-adrenal (HPA) axis, which is the equivalent of the hypothalamus-pituitary-inter-renal (HPI) axis in teleost fish. Both are essential for stress adaptation [1][2][3][4][1,2,3,4]. The axis consists of a highly conserved regulatory system present in all living organisms aimed at maintaining a dynamic equilibrium in the body in response to external and internal stimuli, which is fundamental to assure homeostasis and survival. Cortisol, the end-product of the HPA/I axis, is the main GC both in humans and teleost fish and plays a fundamental role in the maintenance of both resting and stress-related responses [5][6][7][8][5,6,7,8].
Since their discovery in the 1940s [9], much has been learnt of GC molecular modes of action [10][11][12][13][14][15][16][17][18][19][20][21][22][23][10,11,12,13,14,15,16,17,18,19,20,21,22,23]. In particular, the glucocorticoid receptor’s characterization as a DNA-binding protein that regulates transcription initiation [24], the cloning of GR [25][26][25,26] and the breakthrough that most of the immunosuppressive actions of GCs occur via interfering with key inflammatory transcriptional regulators such as NF-κB and AP-1 [27][28][29][30][27,28,29,30], represent the main milestones.
Natural and synthetic glucocorticoids have been widely used for decades as effective anti-inflammatory and immunosuppressive treatments to control pathological disorders, which are very often linked to hypoxia. In particular, they have been broadly used to treat both acute and chronic inflammations, including inflammatory bowel disease, rheumatoid arthritis, multiple sclerosis, eczema and psoriasis, as well as being used in treatment of various leukaemias and in immunosuppressive regimes upon organ transplant [31][32][33][34][35][36][31,32,33,34,35,36]. At any point in time, an estimated ~1% of the total adult UK population receives oral glucocorticoid therapy [37]. However, due to the presence of adverse effects [38] and GC resistance [39][40][41][42][39,40,41,42], their therapeutic benefits are limited in patients chronically treated with these steroids. Examples of the most common GC-related side effects include osteoporosis, glaucoma, diabetes, skin atrophy, abdominal obesity, dyslipidemia, hypertension in adults and growth retardation in children [16][43][44][16,43,44].
Cortisol exerts its functions through direct binding to the glucocorticoid receptor (GR), but also to the mineralocorticoid receptor (MR), which binds cortisol with even higher affinity [45][46][45,46]. As transcription factors, both GR and MR compete for the same ligands, can form heterodimers and homodimers with each other, recognize and bind many of the same hormone response elements on the DNA, and share numerous coregulatory proteins involved in the gene transcription initiation. Importantly, GCs activate MR in most tissues at basal levels, whereas activate GR under stressful conditions or at the diurnal peak [47].
Once bound together, they form an active complex which can function in the nucleus to modulate the transcription of effector proteins, as well as in the cytoplasm to hamper targeted transcription factors activity. Historically, these functions have been coined genomic and non-genomic modes of action, respectively [48][49][50][51][48,49,50,51]. Importantly, GCs and their kindred intracellular receptors, represent critical checkpoints in the endocrine control of vertebrate energy homeostasis. Indeed, if HPA axis activity is not accurately regulated, GC imbalance may result in different pathological conditions such as hypertension, severe cardiovascular, immunological and metabolic complications (e.g., Addison’s disease (GC deficiency) and Cushing’s syndrome (GC excess)) [52][53][54][52,53,54]. In addition, alterations or flaws in the HPA axis response are tightly associated with a broad range of inflammatory and autoimmune diseases, both in humans and in animal models. The latter include Crohn’s disease, rheumatoid arthritis, colitis, inflammatory bowel disease, multiple sclerosis (whose animal equivalent is autoimmune encephalomyelitis), dermatitis, and asthma. Inflammatory conditions include fibromyalgia, chronic fatigue syndrome, depression, and post-traumatic stress disorder (PTSD) [55][56][57][58][59][60][61][55,56,57,58,59,60,61]. Moreover, even if the biological effects induced by GCs are usually adaptive, their abnormal activity may contribute to a series of acute metabolic diseases which include insulin resistance, obesity, and type 2 diabetes [62][63][62,63]. Thus, furthering the research on how GCs precisely work and interact with other pathways may provide better tools to treat these diseases and simultaneously allow the development of selective GR agonists and specific drug-targeting strategies.
Similarly to GCs that are involved in numerous homeostatic maintenance activities (e.g., metabolism of protein, carbohydrate and lipid, etc.) [64], the HIF signalling pathway exerts a pivotal role in ensuring homeostasis, the preservation of which is essential for the correct functioning of the cell. In this regard, the ability to perceive and quickly respond to changes related to environmental oxygen availability is controlled by the hypoxia-inducible factor transcription factors (HIF) family. Hypoxia is a common pathophysiological occurrence, with a profound impact both on human and animal physiology, in which oxygen availability to cells, tissues or to an organ is reduced below a certain threshold (O2 levels < 2%) [65][66][65,66]. HIF transcription factors are key homeostatic regulators which coordinate a metabolic shift from aerobic to anaerobic metabolism to assure cell survival, both in mammals and in zebrafish [67][68][69][70][71][67,68,69,70,71].
The HIF pathway is finely regulated by the PHD3-VHL-E3 ubiquitin ligase complex the aim of which is to maintain low basal HIF levels that can rapidly increase to promptly respond when oxygen levels decrease. This avoids any activation of the HIF pathway under normoxic conditions. As a transcription factor, HIF drives the hypoxic response via binding to specific hypoxia-response elements (HREs). These are involved in decreasing oxygen consumption and increasing oxygen and nutrient delivery [72][73][74][72,73,74]. Interestingly, HIF signalling can tune its own activation via negative feedback by inducing the expression of the oxygen sensors proteins (PHDs), in particular prolyl hydroxylase 3 (PHD3) in zebrafish and PHD2 in humans and mice [75][76][75,76].
However, although the HIF response is aimed at restoring tissue oxygenation and perfusion, it may sometimes be maladaptive and may contribute to the onset of different pathological conditions (e.g., inflammation, stroke, tissue ischemia and growth of solid tumours) [65]. Thus, both glucocorticoids and hypoxia-induced transcriptional responses have been shown to exert crucial roles in tissue homeostasis and in the regulation of cellular responses to stress and inflammation [77][78][79][80][81][82][77,78,79,80,81,82].
2. Glucocorticoids
2.1. Biosynthesis, Secretion and Availability
GCs are essential steroid hormones biosynthesized and secreted by the adrenal cortex/inter-renal gland both in a circadian manner and in response to stress. The latter is generally defined as a status of real or perceived threat to homeostasis. Assuring homeostasis in the presence of stressors requires the activation of an intricate series of coordinated biological responses performed by the nervous, endocrine and immune systems [62][83][62,92]. The key anatomical structures that regulate the stress response are located both in the central nervous system and in peripheral tissues. The primary effectors of the stress response are localized in the paraventricular nucleus (PVN) of the hypothalamus, in the anterior lobe of the pituitary gland and at the level of the adrenal gland. These three main structures are generally referred to as the hypothalamic-pituitary-adrenal (HPA) axis in humans, and as the hypothalamic-pituitary-inter-renal axis (HPI) in zebrafish [62][83][84][62,92,93]. Among these, the hypothalamus is the initial stressor recognition site for both internal and external signals. In mammals, neurons localized in the paraventricular nucleus synthesize both corticotropin-releasing factor (CRF) and arginine vasopressin (AVP), which are released into hypophyseal portal vessels that access the anterior pituitary gland. On the other hand, in teleosts, there is a direct neuronal connection to endocrine cells through the hypophyseal stalk, since they lack a portal system between the hypothalamus and the pituitary gland [85][94]. Here, CRF binding to its receptor localized on pituitary corticotropes triggers the release of adrenocorticotropic hormone (ACTH) into the systemic circulation. In humans, ACTH derives by post translational modification of its precursor encoded by the proopiomelanocortin (POMC) gene. Of note, due to genome duplication, two pomc genes named pomca and pomcb have been identified in zebrafish [86][87][88][95,96,97]. However, only pomca seems to be expressed in the pituitary gland and is required for the inter-renal organ development [89][90][98,99]. The main target of ACTH is the adrenal cortex in humans and the inter-renal tissue in teleosts, where it binds to the melanocortin 2 receptor (MC2R) on the steroidogenic cells. Here, it stimulates cortisol biosynthesis and secretion starting from cholesterol [62][83][91][92][62,92,100,101].
Finally, once released into the systemic circulation, GCs can access target tissues (e.g., liver, heart, and vascular tissues) to exert metabolic and cardiovascular effects and the brain itself, in order to support cognitive processes required to tackle a threatening situation [93][102]. Under non stressful conditions, glucocorticoid levels in the serum are homeostatically controlled by the HPA monitoring activity, whereas glucocorticoid availability is further tuned at a tissue and cellular level. Circulating glucocorticoids are primarily bound to corticosteroid binding globulin (CBG) and just a small percentage (5–15%) is bound to albumin. As a result, the majority of GC is maintained in an inactive form, and only the remaining 5% of systemic GC is free and bioactive. Hence, CBG concentration constitutes a pivotal regulator of cortisol accessibility [94][103].
Importantly, GCs are also able to control their own biosynthesis and secretion by tuning the activity of the HPA/I axis itself. This is particularly important to stop the stress response and avoid an exacerbated reaction [95][104]. This is achieved via a GC-GR mediated negative feedback loop, which acts both at the hypothalamic and anterior pituitary levels, where GC-GR activity inhibits both CRH and POMC (ACTH precursor) biosynthesis and release [2][21][23][96][2,21,23,105]. This occurs via a mechanism that requires GC-GR binding to an nGRE within the pomca promoter [97][106]. For these reasons, pomca is a well-established and frequently used readout of GR activity. In addition, GCs may indirectly control the HPA axis activity through modulation of brain structures activity, such as the amygdala, the hippocampus and the prefrontal cortex, that can, in turn, influence the activity of the paraventricular nucleus [93][98][99][100][102,107,108,109].
2.2. The Glucocorticoid Receptor: Structure and Functions
GCs exert their systemic functions by binding to the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR). Due to their lipophilic nature, GCs can passively diffuse across the plasma membrane into the cytoplasm. Within the cells, their biological availability is then regulated by two enzymes of the 11β-Hydroxysteroid dehydrogenase (11β-HSD) family that work in an opposite fashion. 11β-HSD2 oxidizes cortisol into its inactive form cortisone, reducing GC availability. Vice versa, 11β-HSD1 transforms cortisone to cortisol, thereby increasing local GC activity. Inside the cell, GCs can bind to their specific receptors GR and MR [33][93][101][102][33,102,110,111]. Both receptors, in the absence of their ligands (unbound state) are associated in an inactive oligomeric complex with specific regulatory proteins. Among these, heat shock protein-90 kD (HSP90), which binds both GR and MR to the C-terminal domain, heat shock protein-70 kD (HSP70), p59 immunophilin, Fkbp51 and Fkbp52 and the small p23 phosphoprotein maintain correct protein folding of the receptor [93][103][104][102,112,113]. The GR, which belongs to the nuclear receptor transcription factor family, is composed of different conserved structural domains [105][114]. These include an N-terminal variable region (NTD) required for ligand-independent gene transactivation, which contains a transactivation domain named activation function 1 (AF1). The latter is responsible for the transcriptional activation and is involved in the association with coregulators and the basal transcription machinery. A central DNA-binding domain composed of two zinc fingers has been shown to be crucial both for GR homodimerization and DNA-binding specificity. This is followed by an adjacent flexible hinge region allowing proper DNA binding, dimerization, and nuclear translocation of the receptor [106][115]. Finally, the C-terminal region (LBD) contains the ligand binding domain and a secondary transactivation domain (AF2), regulated by hormone binding, which is essential for dimerization, interaction with cochaperones, coregulators, and other transcription factors [107][108][116,117]. The LBD also comprises a dimer interface which is fundamental for GR function and the binding of the heat shock protein 90 (Hsp 90) [109][118]. Both DBD and LBD include nuclear localization signals, which are required for GR nuclear translocation. Finally, DBD also incorporate the nuclear export signal sequence (NES) which targets it for export from the nucleus to the cytoplasm via the nuclear pore complex [104][113] (Figure 1).

Figure 1. Glucocorticoid receptor domain structure and translational isoforms. The N-terminal domain (NTD), which is required for ligand-independent gene transactivation, includes a transcriptional activation function region (AF1). The latter, which interacts with coregulators and with the basal transcriptional machinery, is the main posttranslational modifications site. The LBD, which is made up of 12 α-helices and 4 β-sheets, forms a hydrophobic pocket needed for GC binding and includes an AF2 domain. The latter allows interaction with coregulators in a ligand-dependent way. Finally, two nuclear localization signals, named NL1 and NL2, are localized in the DBD-hinge region junction and within the LBD, respectively. Asterisks indicate the location of the starting amino acid (aa position: 1, 27, 86, 90, 98, 316, 331, 336) of the eight different GRα translational isoforms, which are characterised by progressively shorter NTDs.
Although the NTD is conserved, literature reviews and sequence alignments of human, monkey, rat, and mouse GRs have revealed that there are another eight conserved AUG start codons in the exon 2 (Figure 1). In humans, these were shown to produce various GR isoforms with progressively shorter N-terminal transactivation domains [104][113]. These are formed due to the presence of alternative Kozak translation initiation sequences which can cause either ribosomal shunting or ribosomal leaky scanning mechanisms. This allows the generation of different GR subtypes with truncated N-termini [110][111][112][113][114][119,120,121,122,123], which are likely to be fully active. This is consistent with data from zebrafish, where a GR mutant line (grsh551), characterized by a 1 bp deletion in the first coding exon (exon 2, Q48fsX3), proved not to have any detectable phenotype [115][124]. This was confirmed further by ISH analysis, which showed that both grsh551 mutants and wildtypes displayed an identical downregulated pomca expression after synthetic GC (Betamethasone 17,21-dipropionate) administration.
Synthetic GR agonists are supposed to trigger a potent GC response, which in turn elicits the GC-GR mediated negative feedback loop, aimed to shut down their own biosynthesis. As previously mentioned, this mainly occurs at the level of the pituitary gland via downregulation of pomca [2][97][102][2,106,111]. For this reason, if GR is not functional, the GC-GR negative feedback loop cannot occur and pomca expression should not be downregulated, as occurs in grsh551 mutants. Indeed, in grsh551 mutants the feedback occurs normally.
In addition to alternative starts, alternative splicing at exon 9 is responsible for generating two different GR splice variants, namely GRα (777 aa) and GRβ (742 aa) [25][116][25,125]. These two receptor isoforms share an identical amino acid sequence between 1–727 aa and then diverge. In particular, the human (h) GRα c C-terminal region contains 50 distinct amino acid residues that form two alpha-helical structures that play a key ligand binding role. In contrast, the hGRβ C-terminal is characterized by a shortened 15 non-homologous, specific amino acid sequence that prevents GC binding [20][117][118][20,126,127]. Hence, hGRβ does not bind traditional glucocorticoid agonists and lacks transactivational activity on GRE-containing promoters, whereas hGRα is the canonical GR isoform. Nevertheless, hGRβ is constitutively present in the nucleus where it has been shown to act as a dominant-negative inhibitor of hGRα’s transactivational properties [118][119][120][127,128,129]. The mechanism behind this inhibition is still uncertain, but several studies have suggested that competition between both hGR isoforms for transcriptional coactivator proteins and/or the formation of inactive GRα-GRβ heterodimers might be responsible for that [117][121][122][123][126,130,131,132].
Moreover, the function of hGRβ extends beyond antagonism of the hGRα isoform [124][133]; for instance, binding to the glucocorticoid antagonist mifepristone (RU486) has been also reported [125][126][134,135]. In addition, it has been shown that increased hGRβ expression is correlated both with the development of immune-related diseases (e.g., ulcerative colitis, leukemia and severe asthma) [127][128][129][136,137,138] and with glucocorticoid resistance in patients affected by these diseases [130][131][132][139,140,141]. Interestingly, previous studies have established the occurrence of a GR β-isoform in zebrafish larvae, which similarly to the hGRβ, confirmed the lack of a role in transcriptional regulation and a dominant-negative inhibitor activity on zGRα [19][20][133][134][19,20,142,143]. In this respect, zebrafish have been shown to be a reliable and useful model system both for GC resistance and glucocorticoid receptor research [2][135][2,85].
2.3. GCs Mechanisms of Action
The conventional view of GC mechanism of action has been recently revised and described as a more complicated multiprotein-regulated process. In this regard, it has been shown that in both humans and zebrafish, upon cortisol binding, GR undergoes a conformational change that involves an FKBP51-FKBP52 exchange. The latter triggers the translocation of the GC-GR active complex into the nucleus. FKBP51 is a cochaperone protein that binds HSP90 and decreases the affinity of GR for cortisol. For this reason, FKBP51 has been considered an inhibitor of GR transcriptional activity and its overexpression has been linked to GC resistance in autoimmune diseases [136][137][138][144,145,146]. After ligand binding, FKBP51 is replaced by FKBP52, which in turn recruits dynein to support translocation of the GC/GR complex to the nucleus [139][140][147,148]. This structural modification exposes the two GR nuclear localisation signals, allowing the hormone-activated GR to dimerize with another GC-GR molecule and to migrate into the nucleus via nuclear pores [141][142][149,150]. Interestingly, this transcription factor complex can also act nongenomically (in the cytoplasm), where it may interact via direct protein-protein interactions with other transcriptional regulators and/or kinases (e.g., basal transcription machinery (BTM); phosphoinositide 3-kinase (PI3K); signal transducer and activator of transcription (STAT)) [63][143][144][145][63,151,152,153] (Figure 2).
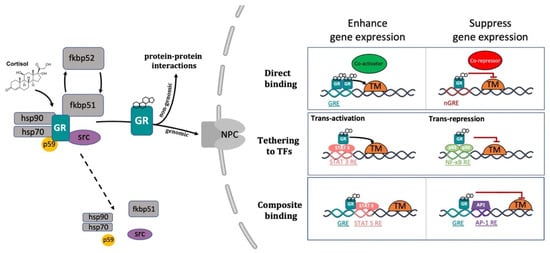
Figure 2. Representative picture of the canonical GR signalling pathway. After binding to GC, GR undergoes a FKBP51-FKBP52 mediated conformational change, becomes hyper-phosphorylated, dissociates from accessory proteins (chaperone complex) and finally translocates into the nucleus. Here, after dimerization with other GRs, it regulates the transcription of target genes by binding to DNA. Interestingly, GR may enhance or repress transcription of target genes by directly binding to palindromic GC response elements (GRE), or by tethering itself to other transcription factors apart from DNA binding, or in a composite manner by both directly binding GRE and interacting with transcription factors bound to neighbouring sites. Created with BioRender.com.
Inside the cell nucleus, GC-GR complexes can directly bind to specific GREs, as tetramers, to upregulate (transactivation) or downregulate (transrepression) the transcription of target genes. Generally, the preferred GRE motif (AGAACAnnnTGTTCT in humans, and GGAACAnnnTGTTCT in zebrafish) is an imperfect palindromic consensus sequence that consists of two 6 bp half sites. The three-nucleotide spacing in-between the two half sites is essential for the GR to tetramerize on this sequence. Previous genome-wide studies have shown that the same GRE can mediate both the GC-dependent induction of many genes (positive GRE) and the repression of others (negative GRE) [104][146][113,154]. Interestingly, the presence of specific inverted repeats negative GREs (IR nGRE), unrelated to simple GREs has also been reported both in mice and in humans. These DNA binding sequences are palindromic sequences consisting of two inverted repeated (IR) motifs separated by 1 bp. In particular, they bind GC-GR complexes to promote the assembly of cis-acting GR-SMRT/NCoR repressing complexes [147][148][155,156] (Figure 2).
In summary, these findings indicate that the broadly different GC effects on various tissues can be partially ascribed to cell type-specific differences in the chromatin landscape that affects the accessibility of specific GREs for GR binding [149][150][157,158]. Furthermore, the GC concentration at which the GR binds to GREs depends on the cell type and chromosomal context. Another important feature of the GC-GR complex that makes its effects even more versatile is that it can tune gene expression in different ways: by binding directly to DNA, by tethering itself to other transcription factors bound to DNA, or via direct binding to DNA and with neighbouring DNA-bound transcriptional regulators (composite manner, Figure 2) [104][151][152][113,159,160].
2.4. The Mineralocorticoid Receptor: Structure and Functions
As previously mentioned, cortisol can bind not only to GR, but also to MR. Both are members of the steroid receptor superfamily (corticosteroid receptors) of ligand-activated transcription factors that enhance or repress the transcription of target genes, as well as promote rapid nongenomic/extra-nuclear events via several cell signalling pathways [47]. The mineralocorticoid receptor is a 984-amino acid cytoplasmic protein that is characterized by three different domains: an N-terminal transcriptional regulator domain, a DNA-binding domain, and a ligand-binding domain responsible for the selectivity of hormone binding. Analogously to GR, in its unliganded state MR is associated with a number of chaperone proteins (HSP90, HSP70, FKBP51 and p23) that play a crucial role in trafficking and maintaining MR in a suitable conformation for ligand binding [153][161].
MR has a ten-fold higher affinity for cortisol than GR and is preferably activated under basal conditions, implying distinct roles for each receptor in the regulation of HPA axis activity [154][155][162,163] It has also been observed that cortisol, even at lower concentrations than those required to activate the GR, binds to MR and might enhance the activity of several kinases (i.e., protein kinase C (PKC), cyclic adenosine 3′,5′-monophosphate (cAMP), and phosphoinositide 3-kinases (PI3K)) involved in different signal transduction cascades [4][47][156][4,47,164]. On the other hand, GR is primarily activated as a result of stress, or at the diurnal peak when circulating cortisol levels are also peaking [154][157][162,165]. Cortisol also exerts broad effects on mood and behaviour via MRs and GRs that are expressed in different regions of the brain [158][159][166,167]. In particular, GR is widely expressed throughout the brain, primarily in the PVN (stress-regulating centre) and in the prefrontal cortex-hippocampal-amygdala circuitry (cognitive, emotional regulation and memory consolidation centre). Vice versa, MR is predominantly expressed in the hippocampus, amygdala, and the lateral septum (emotionality, social behaviour and feeding process hub) [160][168].
In mammals, the mineralocorticoid system is essential to regulate potassium and fluid homeostasis upon aldosterone activation of MR. Even though cortisol is a high-affinity ligand for MR, this steroid is deactivated in specific mineralocorticoid responsive tissues, such as the kidney, by the previously mentioned 11b-HSD-2 enzymatic activity. This allows aldosterone, a second corticosteroid present in mammals, to bind to this receptor. Surprisingly, teleosts do not synthesize aldosterone, and cortisol has been shown to mediate stress axis regulation, as well as the majority of the changes in iono-regulatory and osmo-regulatory functions, via GR and MR signalling [1][4][1,4].
Interestingly, previous work performed both in rats and in teleosts showed that while MR is involved both in basal and onset of stress-induced HPA/I axis activity, GR mainly controls its termination [4][161][4,169]. However, a recent zebrafish study highlighted that rapid locomotor responses to quick changes in light illumination or water salinity (environmental stressors) require GC-GR mediated HPI axis signalling, but not MR [162][170]. Finally, our recently published research suggested that not only GR, but also MR signalling, is involved in the GC-negative feedback regulation (HPI axis termination) and plays a key role in assuring a proper HIF response in teleosts [163][171]. In view of this, furthering the precise role of MR and mineralocorticoid modes of action in vivo, particularly in relation to in the HIF signalling pathway, is warranted.
2.5. The Role of Glucocorticoids in Inflammation
Glucocorticoids suppress most of the events early in the inflammatory response and subsequently facilitate inflammation resolution. They suppress both vasodilation and the enhanced vascular permeability that occurs as a consequence of an inflammatory challenge, and inhibit leukocyte migration from the inflamed region [10][164][165][10,172,173]. In addition, they tune both distribution and trafficking of leukocytes, promote death/survival in some cells and may affect cellular differentiation programmes [165][166][167][168][169][170][173,174,175,176,177,178]. It is well established that many of the anti-inflammatory and immunosuppressive glucocorticoid actions are referable either indirectly or directly to GC-GR mediated transcriptional regulation of numerous genes expressed in leukocytes [165][173]. On the other hand, even though MR expression has been reported in immune cells [171][172][173][179,180,181], its anti-inflammatory role has been considered negligible so far [173][174][181,182], but rather surprisingly, MR-dependent proinflammatory effects have also been noted [175][176][183,184]. It is presently unclear whether these effects are mediated by the glucocorticoid cortisol or the mineralocorticoid aldosterone [177][185].
Glucocorticoids, through GR, can modulate gene expression in different ways [103][108][112,117]. Among these, transactivation is the mechanism by which GC predominantly induce the transcription of numerous anti-inflammatory genes, such as GILZ, MKP-1 and IĸBα. This occurs through direct binding of single or multiple GC-GRs to palindromic glucocorticoid response elements (GREs) [178][179][180][181][186,187,188,189]. Importantly, this mode of action has also been shown to be responsible for several undesirable metabolic side effects linked to chronic GC treatment [140][148]. On the other hand, transrepression is the mechanism by which GC downregulate the transcription of inflammatory genes and requires direct protein-protein interaction of GR to other transcription factors. This mode of action is generally accepted to convey the beneficial GC anti-inflammatory effects, which are mainly implicated in rapid cellular responses [40][140][182][183][184][40,148,190,191,192]. In particular, transrepression is known to mainly occur via direct binding between the monomeric GC-GR complex and transcription factors (e.g., AP-1, NF-kB, c-Jun, and c-Fos) activated by cytokines and other pro-inflammatory stimuli, which synergistically coordinate the expression of several proinflammatory genes [27][100][152][185][186][187][188][27,109,160,193,194,195,196]. Of note, most of these genes are commonly overexpressed during chronic nonresolving inflammatory states. Interestingly, transrepression is not restricted to these transcription factors but also includes others such as CREB, STAT, and T-bet [152][189][160,197]. As a result, the mutual antagonism between transcription factors frequently impairs their transcriptional properties and prevents them from binding to their corresponding DNA response elements.
As a consequence of the above considerations, optimal GC analogs should be characterized by a high inhibitory activity against inflammatory mediators, coupled with a low transactivation activity, in order to induce minimal side effects. Interestingly, different steroidal and nonsteroidal ligands have been reported to have this dual function (e.g., RU-24858 and ZK-216348) [140][183][190][191][192][148,191,198,199,200]. These compounds have been reported to suppress key inflammatory and immune transcription factor activity in vivo [190][191][192][193][194][198,199,200,201,202]. However, as stated before, since GCs can trigger gene expression via multiple routes, unexpected secondary side effects might occur. For this reason, further research is also warranted to elucidate the implications of the nongenomic GC-mediated activity both in the immune and inflammatory scenario.
Importantly, although the GC-GR complex is known for its anti-inflammatory effects, the picture is more complex. Indeed, contrary to expectations, GR loss of function was reported by Facchinello and coworkers to prevent the transcriptional activity linked to the inflammatory immune response (i.e., of cytokines Il6, Il1β, Il8 and Mmp-13) [8], corroborating the hypothesis of a GC-GR mediated dual action on the immune system [35][195][35,203]. However, it is clear that further research is warranted to better elucidate this aspect. In addition, GR was shown to synergistically induce proinflammatory genes by acting on other signalling pathways [196][197][198][199][204,205,206,207]. Finally, studies also demonstrated that GCs increase the transcription of numerous anti-inflammatory molecules such as interleukin-10 (IL-10), interleukin-1 receptor antagonist (IL-1RA), secretory leukocyte inhibitory protein and neutral endopeptidase [41][200][41,208].
Previous research also revealed that alterations in chromatin structure are important for modulating the outcome of GC activity. Indeed, GR can interact differently with histone acetyltransferases (HATs), with histone deacetylases (HDACs) and also kinases (i.e., MSK1, PKA and JNK) [152][160]. These may, in turn, modulate the chromatin environment by modifying chromatin accessibility and further tuning inflammatory and immune gene expression [152][160]. Furthermore, since chromatin accessibility can predetermine GR binding patterns and is crucial for cell-specific outcomes, it can provide novel molecular basis for tissue selectivity [201][202][209,210]. In addition, another study showed that GR may directly inhibit CREB binding protein (CBP)-associated HAT activity and may recruit HDAC2 to the p65-CBP HAT complex [203][211]. This novel glucocorticoid repression mechanism suggests that histone acetylation inhibition represents an additional level of control of inflammatory gene expression. Consequently, this further indicates that pharmacologically manipulating specific histone acetylation status could be an alternative approach for treating inflammatory diseases.