 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Danuta Szkutnik-Fiedler | + 3181 word(s) | 3181 | 2021-12-17 04:43:52 | | | |
| 2 | Beatrix Zheng | + 52 word(s) | 3233 | 2021-12-23 02:23:37 | | |
Video Upload Options
A combination of the tyrosine kinase inhibitor—sorafenib—and the opioid analgesic—morphine—can be found in the treatment of cancer patients. Since both are substrates of P-glycoprotein (P-gp), and sorafenib is also an inhibitor of P-gp, their co-administration may affect their pharmacokinetics, and thus the safety and efficacy of cancer therapy.
1. Introduction
2. Current Insights
2.1. The Influence of Morphine on the Pharmacokinetics of Sorafenib and SR_NO
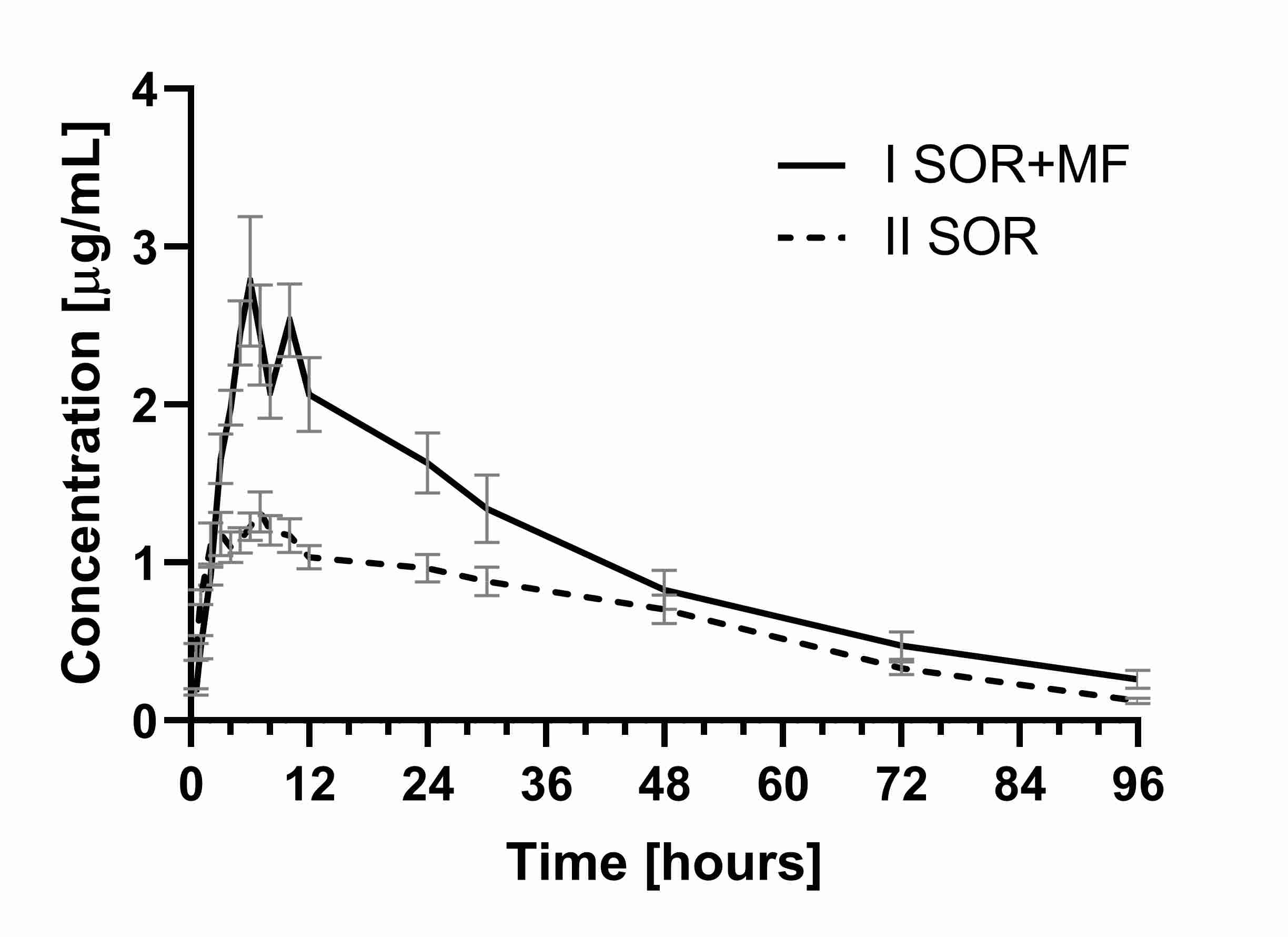
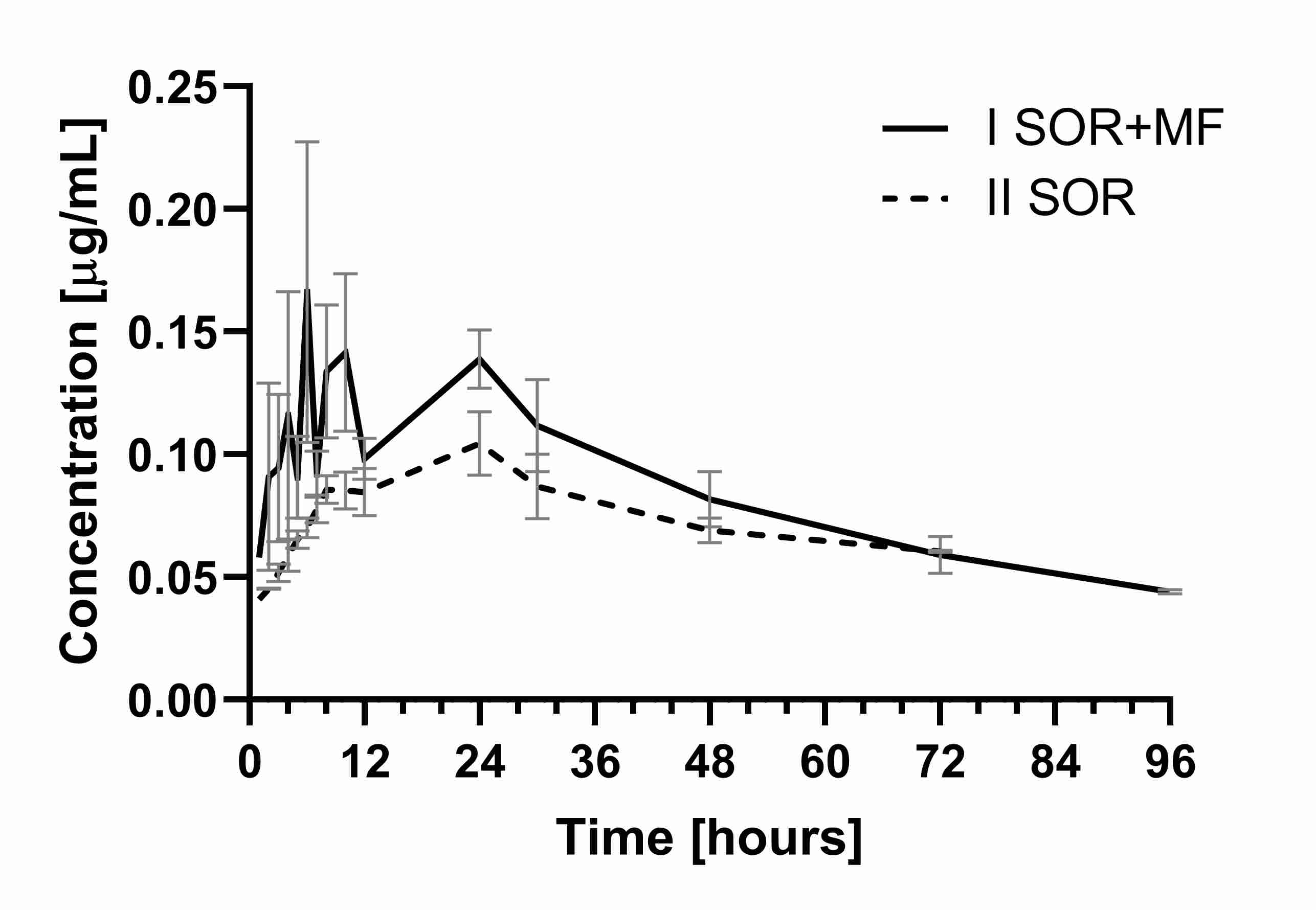
2.2. The Influence of Sorafenib on the Pharmacokinetics of Morphine and M3G
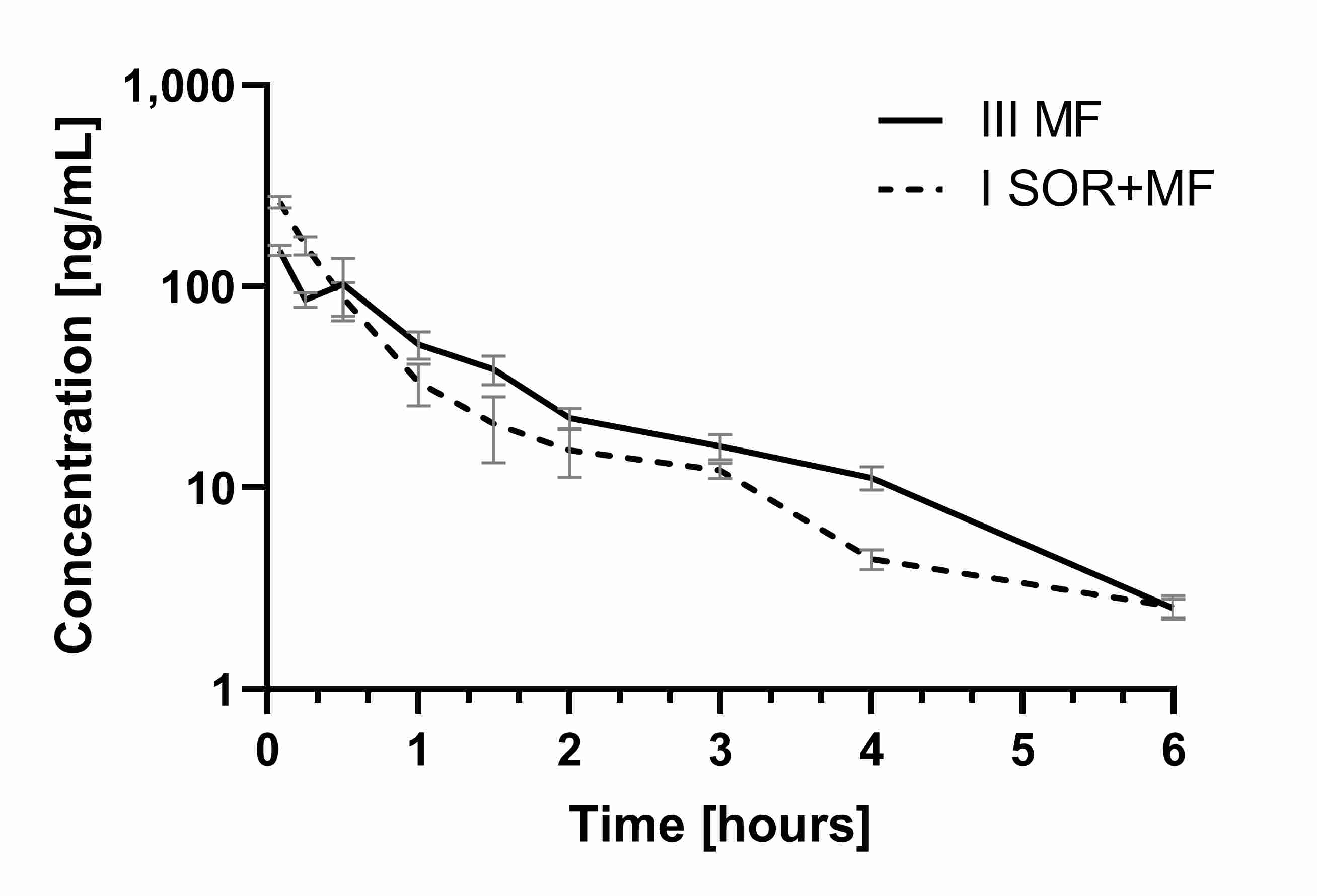
Table 1. Pharmacokinetic parameters of sorafenib and its metabolite sorafenib N-oxide after a single oral dose of100 mg/kg b.w. of sorafenib (IISOR group) and a single oral dose of 100 mg/kg b.w. of sorafenib + single intraperitoneal dose 5 mg/kg b.w. of morphine (ISOR+MF group).
|
Pharmacokinetic Parameters |
IISOR (n = 8) |
ISOR+MF (n = 8) |
p-Value ISOR+MF vs. IISOR |
Gmean Ratio * (90% CI) ISOR+MF vs. IISOR |
|
sorafenib |
||||
|
Cmax (µg/mL) |
1.56 ± 0.35 (22.6) |
3.25 ± 0.80 (24.6) |
0.0030 |
2.08 (1.70; 2.55) |
|
Cmax/(D/kg) |
0.03 ± 0.01 (23.9) |
0.07 ± 0.02 (23.6) |
<0.0001 |
2.13 (1.73; 2.62) |
|
AUC0–t (µg × h/mL) |
62.83 ± 16.14 (25.7) |
97.98 ± 30.17 (30.8) |
0.0115 |
1.53 (1.16; 2.02) |
|
AUC0–t/(D/kg) (µg × h × kg/mL/mg) |
1.25 ± 0.32 (25.4)
|
1.99 ± 0.59 (29.7) |
0.0075 |
1.57 (1.20; 2.05) |
|
AUC0–∞ (µg × h/mL) |
67.05 ± 16.70 (24.9) |
109,08 ± 37.18 (34.1) |
0.0115 |
1.58 (1.17; 2.13) |
|
AUC0–∞/(D/kg) (µg × h × kg/mL/mg) |
1.33 ± 0.33 (24.9) |
2.21 ± 0.73 (32.9) |
0.0076 |
1.62 (1.21; 2.16) |
|
tmax (h) |
5.13 ± 2.17 (42.3) |
7.63 ± 2.72 (35.7) |
0.0616 |
1.54 (1.06; 2.25) |
|
ka (h−1) |
0.74 ± 0.31 (42.5) |
0.03 ± 0.01 (20.5) |
0.0008 |
0.03 (0.03; 0.05) |
|
kel (h−1) |
0.035 ± 0.01 (30.3) |
0.29 ± 0.16 (53.5) |
0.0008 |
8.16 (5.98; 11.12) |
|
t1/2 (h) |
21.89 ± 7.79 (35.6) |
27.31 ± 5.32 (19.5) |
0.0829 |
1.25 (1.02; 1.53) |
|
Cl/F (L/h × kg) |
0.80 ± 0.22 (27.1) |
0.51 ± 0.23 (43.8) |
0.0224 |
0.62 (0.46; 0.83) |
|
Vd/F (L) |
25.30 ± 11.59 (45.8) |
19.14 ± 5.26 (27.5) |
0.0829 |
0.77 (0.59; 1.01) |
|
sorafenib N-oxide |
||||
|
Cmax (µg/mL) |
0.11 ± 0.02 (21.8) |
0.27 ± 0.16 (57.5) |
0.0022 |
2.15 (1.50; 3.08) |
|
AUC0–t (µg × h/mL) |
4.10 ± 1.56 (38.1) |
6.64 ± 2.44 (36.8) |
0.0268 |
1.64 (1.14; 2.36) |
|
AUC0–∞ (µg × h/mL) |
8.61 ± 2.19 (25.4) |
9.39 ± 2.97 (31.6) |
0.1242 |
1.41 (0.95; 2.08) |
|
tmax (h) |
16.38 ± 8.21 (50.1) |
14.50 ± 11.40 (78.6) |
0.4531 |
0.76 (0.41; 1.39) |
|
kel (h−1) |
0.016 ± 0.010 (60.9) |
0.023 ± 0.012 (51.6) |
0.4001 |
0.80 (0.47; 1.36) |
|
t1/2 (h) |
53.31 ± 25.23 (47.3) |
39.30 ± 22.54 (57.4) |
0.4622 |
1.25 (0.73; 2.13) |
|
ratio sorafenib N-oxide/sorafenib |
||||
|
Cmax (µg/mL) |
0.07 ± 0.02 (26.8) |
0.09 ± 0.07 (76.2) |
0.5280 |
1.03 (0.67; 1.61) |
|
AUC0–t (µg × h/mL) |
0.07 ± 0.02 (37.1) |
0.07 ± 0.03 (43.7) |
0.6012 |
1.07 (0.70; 1.65) |
|
AUC0–∞ (µg × h/mL) |
0.14 ± 0.05 (38.1) |
0.10 ± 0.07 (64.1) |
0.9176 |
0.89 (0.53; 1.51) |
Cmax, maximum observed plasma concentration; AUC0–t, area under the plasma concentration-time curve from zero to the last measurable concentration; AUC0–∞, area under the plasma concentration-time curve from zero to infinity; tmax, time to reach the Cmax; ka, absorption rate constant; kel, elimination rate constant; t1/2, half-life in the elimination phase; Cl/F, apparent plasma drug clearance; Vd/F, apparent volume of distribution; b.w., body weight; the pharmacokinetic parameter values are shown as the arithmetic means ± standard deviations (SD) with coefficients of variation (CV) (%) in the brackets; * geometric means (Gmean) ratio between ISOR+MF and IISOR groups (%) with a 90% confidence interval (CI) in the brackets; individual drug ratios were calculated according the following equations: the metabolite Cmax (ng/mL)/parent Cmax (ng/mL), metabolite AUC0–t (ng × h/mL)/parent drug AUC0–t (ng × h/mL), and metabolite AUC0–∞ (ng × h/mL)/parent drug AUC0–∞ (ng × h/mL).
Table 2. Pharmacokinetic parameters of morphine and its metabolite M3G after a single intraperitoneal dose of 5 mg/kg b.w. of morphine (IIIMF group) and a single oral dose of 100 mg/kg b.w. of sorafenib + single intraperitoneal dose of 5 mg/kg b.w. of morphine (ISOR+MF group).
|
Pharmacokinetic Parameters |
IIIMF (n = 7) |
ISOR+MF (n = 8) |
p-Value ISOR+MF vs. IIIMF |
Gmean Ratio * (90% CI) ISOR+MF vs. IIIMF |
|
morphine |
||||
|
Cmax (ng/mL) |
166.83 ± 46.12 (27.6) |
261.83 ± 47.85 (18.3) |
0.0018 |
1.59 (1.30; 1.94) |
|
Cmax/(D/kg) (kg × ng/mL/mg) |
67.88 ± 18.74 (27.6) |
106.14 ± 18.96 (17.9) |
0.0018 |
1.58 (1.30; 1.93) |
|
AUC0–t (ng × h/mL) |
169.88 ± 47.37 (27.9) |
155.40 ± 28.41 (18.3) |
0.4786 |
0.93 (0.75; 1.16) |
|
AUC0–t/(D/kg) (ng × h × kg/mL/mg) |
69.28 ± 19.99 (28.9) |
63.06 ± 11.52 (18.3) |
0.4655 |
0.93 (0.75; 1.16) |
|
AUC0–∞ (ng × h/mL) |
174.44 ± 46.73 (26.8) |
162.57 ± 25.89 (15.9) |
0.5458 |
0.95 (0.78; 1.17) |
|
AUC0–∞/(D/kg) (ng × h × kg/mL/mg) |
71.14 ± 19.75 (27.8) |
65.96 ± 10.49 (15.9) |
0.5287 |
0.95 (0.77; 1.17) |
|
tmax (h) |
0.14 ± 0.16 (110.2) |
0.08 ± 0.00 (0.0) |
0.3559 |
0.77 (0.47; 1.27) |
|
kel (h−1) |
0.60 ± 0.18 (29.3) |
0.54 ± 0.41 (76.1) |
0.2976 |
0.75 (0.46; 1.23) |
|
t1/2 (h) |
1.22 ± 0.28 (22.7) |
1.91 ± 1.13 (59.3) |
0.2976 |
1.33 (0.82; 2.15) |
|
Cl/F (L/h × kg) |
15.05 ± 4.39 (29.2) |
15.51 ± 2.36 (15.2) |
0.8010 |
1.06 (0.86; 1.30) |
|
Vd/F (L/kg) |
27.49 ± 13.38 (48.7) |
44.65 ± 29.20 (65.4) |
0.1778 |
1.40 (0.76; 2.60) |
|
M3G |
||||
|
Cmax (ng/mL) |
9781.28 ± 3184.17 (32.6) |
20,796.81 ± 3657.84,(17.6) |
<0.0001 |
2.20 (1.73; 2.80) |
|
AUC0–t (ng × h/mL) |
10,035.88 ± 1408.48 (14.0) |
14,734.53 ± 3979.45 (27.0) |
0.0124 |
1.43 (1.14; 1.79) |
|
AUC0–∞ (ng × h/mL) |
10,131.53 ± 1393.15 (13.8) |
14,871.32 ± 4001.35 (26.9) |
0.0121 |
1.42 (1.14; 1.79) |
|
tmax (h) |
0.46 ± 0.09 (20.4) |
0.34 ± 0.13 (37.6) |
0.0662 |
0.71 (0.53; 0.96) |
|
kel (h−1) |
0.73 ± 0.20 (27.6) |
0.68 ± 0.19 (28.0) |
0.6355 |
0.93 (0.69; 1.24) |
|
t1/2 (h) |
1.03 ± 0.33 (32.5) |
1.13 ± 0.47 (41.1) |
0.7285 |
1.08 (0.81; 1.45) |
|
M3G/morphine |
||||
|
Cmax (ng/mL) |
63.87 ± 30.71 (48.1) |
82.18 ± 21.67 (26.4) |
0.2004 |
1.14 (0.78; 1.66) |
|
AUC0–t (ng × h/mL) |
64.45 ± 24.06 (37.3) |
97.35 ± 33.05 (34.0) |
0.0488 |
1.38 (0.96; 2.00) |
|
AUC0–∞ (ng × h/mL) |
62.88 ± 22.44 (35.7) |
92.80 ± 28.44 (30.7) |
0.0435 |
1.53 (1.10; 2.12) |
Cmax, maximum observed plasma concentration; AUC0–t, area under the plasma concentration-time curve from zero to the last measurable concentration; AUC0–∞, area under the plasma concentration-time curve from zero to infinity; tmax, time to reach the Cmax; ka, absorption rate constant; kel, elimination rate constant; t1/2, half-life in the elimination phase; Cl/F, apparent plasma drug clearance; Vd/F, apparent volume of distribution; b.w., body weight; the pharmacokinetic parameter values are shown as the arithmetic means ± standard deviations (SD) with coefficients of variation (CV) (%) in the brackets; * geometric means (Gmean) ratio between ISOR+MF and IISOR groups (%) with a 90% confidence interval (CI) in the brackets; individual drug ratios were calculated according the following equations: the metabolite Cmax (ng/mL)/parent Cmax (ng/mL), metabolite AUC0–t (ng × h/mL)/parent drug AUC0–t (ng × h/mL), and metabolite AUC0–∞ (ng × h/mL)/parent drug AUC0–∞ (ng × h/mL).
References
- Gress, K.L.; Charipova, K.; Kaye, A.D.; Viswanath, O.; Urits, I. An Overview of Current Recommendations and Options for the Management of Cancer Pain: A Comprehensive Review. Oncol. Ther. 2020, 8, 251–259.
- Karbownik, A.; Miedziaszczyk, M.; Grabowski, T.; Stanisławiak-Rudowicz, J.; Jaźwiec, R.; Wolc, A.; Grześkowiak, E.; Szałek, E. In vivo assessment of potential for UGT-inhibition-based drug-drug interaction between sorafenib and tapentadol. Biomed. Pharmacother. 2020, 130, 110530.
- Gong, L.; Giacomini, M.M.; Giacomini, C.; Maitland, M.L.; Altman, R.B.; Klein, T.E. PharmGKB summary: Sorafenib pathways. Pharm. Genom. 2017, 27, 240–246.
- Bertot, L.C.; Adams, L.A. Trends in hepatocellular carcinoma due to non-alcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 179–187.
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009.
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795.
- Miners, J.O.; Chau, N.; Rowland, A.; Burns, K.; MnKinnon, R.A.; Mackenzie, P.I.; Tucker, G.T.; Knights, K.M.; Kichenadasse, G. Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: Implications for hyperbilirubinemia. Biochem. Pharmacol. 2017, 129, 85–95.
- Chen, M.; Neul, C.; Schaeffeler, E.; Frisch, F.; Winter, S.; Schwab, M.; Koepsell, H.; Hu, S.; Laufer, S.; Baker, S.D.; et al. Sorafenib activity and disposition in liver cancer does not depend on organic cation transporter 1. Clin. Pharmacol. Ther. 2020, 107, 227–237.
- Vasilyeva, A.; Durmus, S.; Li, L.; Wagenaar, E.; Hu, S.; Gibson, A.A.; Panetta, J.C.; Mani, S.; Sparreboom, A.; Baker, S.D.; et al. Hepatocellular shuttling and recirculation of sorafenib-glucuronide is dependent on Abcc2, Abcc3, and Oatp1a/1b. Cancer Res. 2015, 75, 2729–2736.
- Summary of Product Characteristic for Nexavar 200 mg, European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/product-information/nexavar-epar-product-information_en.pdf (accessed on 6 April 2021).
- Karbownik, A.; Stachowiak, A.; Urjasz, H.; Sobańska, K.; Szczecińska, A.; Grabowski, T.; Stanisławiak-Rudowicz, J.; Wolc, A.; Grześkowiak, E.; Szałek, E. The oxidation and hypoglycaemic effect of sorafenib in streptozotocin-induced diabetic rats. Pharmacol. Rep. 2020, 72, 254–259.
- Wang, X.; Zhang, X.; Huang, X.; Li, Y.; Wu, M.; Liu, J. The drug-drug interaction of sorafenib mediated by P-glicoprotein and CYP3A4. Xenobiotica 2015, 46, 651–658.
- Kimura, Y.; Shibata, M.; Tamada, M.; Ozaki, N.; Arai, K. Pharmacokinetics of Morphine in Rats with Adjuvant-induced Arthritis. In Vivo 2017, 31, 811–817.
- Gadeyne, C.; Van der Heyden, S.; Gasthuys, F.; Croubels, S.; Schauvliege, S.; Polis, I. The influence of modulation of P-glycoprotein and/or cytochrome P450 3A on the pharmacokinetics and pharmacodynamics of orally administered morphine in dogs. J. Vet. Pharmacol. Ther. 2011, 34, 417–423.
- De Gregori, S.; De Gregori, M.; Ranzani, G.N.; Allegri, M.; Minella, C.; Regazzi, M. Morphine metabolism, transport and brain disposition. Metab. Brain Dis. 2012, 27, 1–5.
- Ing Lorenzini, K.; Girardin, F. Direct-acting antiviral interactions with opioids, alcohol or illicit drugs of abuse in HCV-infected patients. Liver Int. 2020, 40, 32–44.
- Schinkel, A.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; Borst, P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J. Clin. Investig. 1995, 96, 1698–1705.
- Letrent, S.P.; Polli, J.W.; Humphreys, J.E.; Pollack, G.M.; Brouwer, K.R.; Brouwer, K.L. P-glycoprotein-mediated transport of morphine in brain capillary endothelial cells. Biochem. Pharmacol. 1999, 58, 951–957.
- Crowe, A. The influence of P-glycoprotein on morphine transport in Caco-2 cells. Comparison with paclitaxel. Eur. J. Pharmacol. 2002, 440, 7–16.
- Xie, R.; Hammarlund-Udenaes, M.; de Boer, A.G.; de Lange, E.C. The role of P-glycoprotein in blood-brain barrier transport of morphine: Transcortical microdialysis studies in mdr1a (-/-) and mdr1a (+/+) mice. Br. J. Pharmacol. 1999, 128, 563–568.
- Letrent, S.P.; Pollack, G.M.; Brouwer, K.R.; Brouwer, K.L. Effect of GF120918, a potent P-glycoprotein inhibitor, on morphine pharmacokinetics and pharmacodynamics in the rat. Pharm Res. 1998, 15, 599–605.
- Aquilante, C.L.; Letrent, S.P.; Pollack, G.M.; Brouwer, K.L. Increased brain P-glycoprotein in morphine tolerant rats. Life Sci. 2000, 66, PL47–PL51.
- Fujita-Hamabe, W.; Nishida, M.; Nawa, A.; Kobori, T.; Nakamoto, K.; Kishioka, S.; Tokuyama, S. Etoposide modulates the effects of oral morphine analgesia by targeting the intestinal P-glycoprotein. J. Pharm. Pharmacol. 2012, 64, 496–504.
- Seleman, M.; Chapy, H.; Cisternino, S.; Courtin, C.; Smirnova, M.; Schlatter, J.; Chiadmi, F.; Scherrmann, J.M.; Noble, F.; Marie-Claire, C. Impact of P-glycoprotein at the blood-brain barrier on the uptake of heroin and its main metabolites: Behavioral effects and consequences on the transcriptional responses and reinforcing properties. Psychopharmacology 2014, 231, 3139–3149.
- Zong, J.; Pollack, G.M. Morphine antinociception is enhanced in mdr1a gene-deficient mice. Pharm. Res. 2000, 17, 749–753.
- Fudin, J.; Fontenelle, D.V.; Payne, A. Rifampin reduces oral morphine absorption: A case of transdermal buprenorphine selection based on morphine pharmacokinetics. J. Pain Palliat. Care Pharmacother. 2012, 26, 362–367.
- Wang, J.; Cai, B.; Huang, D.X.; Yang, S.D.; Guo, L. Decreased analgesic effect of morphine, but not buprenorphine, in patients with advanced P-glycoprotein(+) cancers. Pharmacol. Rep. 2012, 64, 870–877.
- Meissner, K.; Avram, M.J.; Yermolenka, V.; Francis, A.M.; Blood, J.; Kharasch, E.D. Cyclosporine-inhibitable blood-brain barrier drug transport influences clinical morphine pharmacodynamics. Anesthesiology 2013, 119, 941–953.
- Drewe, J.; Ball, H.A.; Beglinger, C.; Peng, B.; Kemmler, A.; Schächinger, H.; Haefeli, W.E. Effect of P-glycoprotein modulation on the clinical pharmacokinetics and adverse effects of morphine. Br. J. Clin. Pharmacol. 2000, 50, 237–246.
- Lötsch, J.; Schmidt, R.; Vetter, G.; Schmidt, H.; Niederberger, E.; Geisslinger, G.; Tegeder, I. Increased CNS uptake and enhanced antinociception of morphine-6-glucuronide in rats after inhibition of P-glycoprotein. J. Neurochem. 2002, 83, 241–248.
- Gharavi, R.; Hedrich, W.; Wang, H.; Hassan, H.E. Transporter-Mediated Disposition of Opioids: Implications for Clinical Drug Interactions. Pharm. Res. 2015, 32, 2477–2502.
- Thompson, S.J.; Koszdin, K.; Bernards, C.M. Opiate-induced analgesia is increased and prolonged in mice lacking P-glycoprotein. Anesthesiology 2000, 92, 1392–1399.
- Dagenais, C.; Zong, J.; Ducharme, J.; Pollack, G.M. Effect of mdr1a P-glycoprotein gene disruption, gender, and substrate concentration on brain uptake of selected compounds. Pharm. Res. 2001, 18, 957–963.
- Hamabe, W.; Maeda, T.; Kiguchi, N.; Yamamoto, C.; Tokuyama, S.; Kishioka, S. Negative relationship between morphine analgesia and P-glycoprotein expression levels in the brain. J. Pharmacol. Sci. 2007, 105, 353–360.
- Fudin, J.; Fontenelle, D.V.; Fudin, H.R.; Carlyn, C.; Hinden, D.A.; Ashley, C.C. Potential P-glycoprotein pharmacokinetic interaction of telaprevir with morphine or methadone. J. Pain Palliat. Care Pharmacother. 2013, 27, 261–267.
- Tournier, N.; Declèves, X.; Saubaméa, B.; Scherrmann, J.M.; Cisternino, S. Opioid transport by ATP-binding cassette transporters at the blood-brain barrier: Implications for neuropsychopharmacology. Curr. Pharm. Des. 2011, 17, 2829–2842.
- Wandel, C.; Kim, R.; Wood, M.; Wood, A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology 2002, 96, 913–920.
- Tzvetkov, M.V.; dos Santos Pereira, J.N.; Meineke, I.; Saadatmand, A.R.; Stingl, J.C.; Brockmöller, J. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem. Pharmacol. 2013, 86, 666–678.
- Venkatasubramanian, R.; Fukuda, T.; Niu, J.; Mizuno, T.; Chidambaran, V.; Vinks, A.A.; Sadhasivam, S. ABCC3 and OCT1 genotypes influence pharmacokinetics of morphine in children. Pharmacogenomic 2014, 15, 1297–1309.
- Van de Wetering, K.; Zelcer, N.; Kuil, A.; Feddema, W.; Hillebrand, M.; Vlaming, M.L.; Schinkel, A.H.; Beijnen, J.H.; Borst, P. Multidrug resistance proteins 2 and 3 provide alternative routes for hepatic excretion of morphine-glucuronides. Mol. Pharmacol. 2007, 72, 387–394.
- Bourasset, F.; Cisternino, S.; Temsamani, J.; Scherrmann, J.M. Evidence for an active transport of morphine-6-beta-d-glucuronide but not P-glycoprotein-mediated at the blood-brain barrier. J. Neurochem. 2003, 86, 1564–1567.
- Van Leeuwen, R.W.F.; Van Gelder, T.; Mathijssen, R.H.J.; Jansman, F.G.A. Drug-drug interactions with tyrosine-kinase inhibitors: A clinical perspective. Lancet Oncol. 2014, 15, 315–326.
- Yu, J.; Petrie, I.D.; Levy, R.H.; Ragueneau-Majlessi, I. Mechanisms and Clinical Significance of Pharmacokinetic-Based Drug-Drug Interactions with Drugs Approved by the U.S. Food and Drug Administration in 2017. Drug Metab. Dispos. 2019, 47, 135–144.
- Peng, Y.; Cheng, Z.; Xie, F. Evaluation of Pharmacokinetic Drug-Drug Interactions: A Review of the Mechanisms, In Vitro and In Silico Approaches. Metabolites 2021, 11, 75.
- In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Guidance for Industry. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). January 2020, Clinical Pharmacology. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 8 November 2021).
- Cole, S.; Kerwash, E.; Andersson, A. A summary of the current drug interaction guidance from the European Medicines Agency and considerations of future updates. Drug Metab. Pharmacokinet. 2020, 35, 2–11.
- Azam, C.; Claraz, P.; Chevreau, C.; Vinson, C.; Cottura, E.; Mourey, L.; Pouessel, D.; Guibaud, S.; Pollet, O.; Le Goff, M.; et al. Association between clinically relevant toxicities of pazopanib and sunitinib and the use of weak CYP3A4 and P-gp inhibitors. Eur. J. Clin. Pharmacol. 2020, 76, 579–587.
- Oostendorp, R.L.; Buckl, T.; Beijnen, J.H.; van Tellingen, O.; Schellens, J.H. The effect of P-gp (Mdr1a/1b), BCRP (Bcrp1) and P-gp/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Investig. New Drugs 2009, 27, 31–40.
- Teo, Y.L.; Ho, H.K.; Chan, A. Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: Current understanding, challenges and recommendations. Br. J. Clin. Pharmacol. 2015, 79, 241–253.
- Campa, D.; Gioia, A.; Tomei, A.; Poli, P.; Barale, R. Association of ABCB1/MDR1 and OPRM1 gene polymorphisms with morphine pain relief. Clin. Pharmacol. Ther. 2008, 83, 559–566.
- Coulbault, L.; Beaussier, M.; Verstuyft, C.; Weickmans, H.; Dubert, L.; Trégouet, D.; Descot, C.; Parc, Y.; Lienhart, A.; Jaillon, P.; et al. Environmental and genetic factors associated with morphine response in the postoperative period. Clin. Pharmacol. Ther. 2006, 79, 316–324.
- Hasegawa, Y.; Kishimoto, S.; Shibatani, N.; Nomura, H.; Ishii, Y.; Onishi, M.; Inotsume, N.; Takeuchi, Y.; Fukushima, S. The pharmacokinetics of morphine and its glucuronide conjugate in a rat model of streptozotocin-induced diabetes and the expression of MRP2, MRP3 and UGT2B1 in the liver. J. Pharm. Pharmacol. 2010, 62, 310–314.

