 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Weimin Zhang | + 3384 word(s) | 3384 | 2021-12-22 07:46:17 | | | |
| 2 | Conner Chen | Meta information modification | 3384 | 2021-12-23 02:32:46 | | |
Video Upload Options
Gliotoxin is a kind of epipolythiodioxopiperazine derived from different fungi that is characterized by a disulfide bridge. Gliotoxins can be biosynthesized by a gli gene cluster and regulated by a positive GliZ regulator. Gliotoxins show cytotoxic effects via the suppression the function of macrophage immune function, inflammation, antiangiogenesis, DNA damage by ROS production, peroxide damage by the inhibition of various enzymes, and apoptosis through different signal pathways. In the other hand, gliotoxins can also be beneficial with different doses. Low doses of gliotoxin can be used as an antioxidant, in the diagnosis and treatment of HIV, and as an anti-tumor agent in the future. Gliotoxins have also been used in the control of plant pathogens, including Pythium ultimum and Sclerotinia sclerotiorum.
1. The Biosynthesis of Gliotoxin
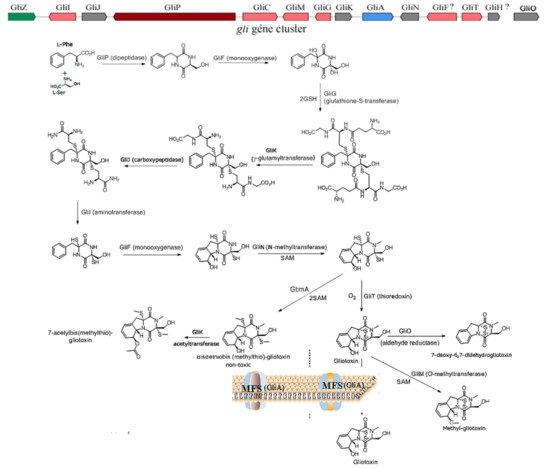
2. The Underlying Toxic Mechanism of Gliotoxin
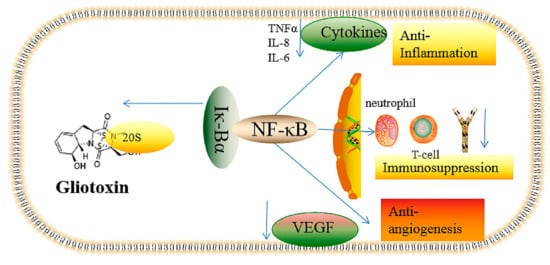
2.1. The Inhibition of Angiogenesis
2.2. Immunosuppressive Activity
2.3. Inflammatory and Anti-Inflammatory Effects
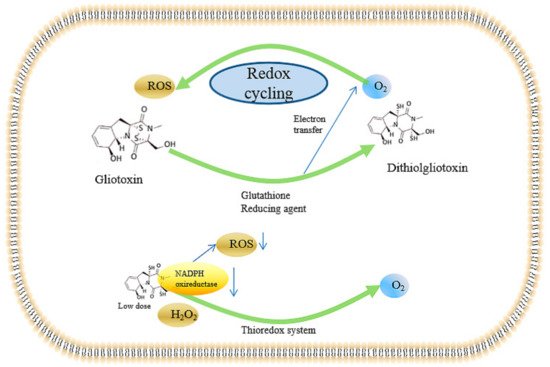
2.4. Inducing the Production of ROS and the Inhibition of Peroxidase
2.5. Genotoxicity of Gliotoxin
References
- Singh, S.; Dureja, P.; Tanwar, R.; Singh, A. Production and antifungal activity of secondary metabolites of Trichoderma virens. Pestic. Res. J. 2005, 17, 26–29.
- Kupfahl, C.; Michalka, A.; Lass-Flörl, C.; Fischer, G.; Haase, G.; Ruppert, T.; Geginat, G.; Hof, H. Gliotoxin production by clinical and environmental Aspergillus fumigatus strains. Int. J. Med. Microbiol. Suppl. 2008, 298, 319–327.
- Lewis, R.E.; Wiederhold, N.P.; Lionakis, M.S.; Prince, R.A.; Kontoyiannis, D.P. Frequency and Species Distribution of Gliotoxin-Producing Aspergillus Isolates Recovered from Patients at a Tertiary-Care Cancer Center. J. Clin. Microbiol. 2005, 43, 6120–6122.
- Scharf, D.H.; Heinekamp, T.; Remme, N.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. Biosynthesis and function of gliotoxin in Aspergillus fumigatus. Appl. Microbiol. Biotechnol. 2011, 93, 467–472.
- Dolan, S.K.; O’Keeffe, G.; Jones, G.W.; Doyle, S. Resistance is not futile: Gliotoxin biosynthesis, functionality and utility. Trends Microbiol. 2015, 23, 419–428.
- Bok, J.W.; Chung, D.; Balajee, S.A.; Marr, K.A.; Andes, D.; Nielsen, K.F.; Frisvad, J.C.; Kirby, K.A.; Keller, N.P. GliZ, a tran-scriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect. Immun. 2006, 74, 6761–6768.
- Bok, J.W.; Keller, N.P. LaeA, a Regulator of Secondary Metabolism in Aspergillus spp. Eukaryot. Cell 2004, 3, 527–535.
- Dhingra, S.; Andes, D.; Calvo, A.M. VeA Regulates Conidiation, Gliotoxin Production, and Protease Activity in the Opportunistic Human Pathogen Aspergillus fumigatus. Eukaryot. Cell 2012, 11, 1531–1543.
- Ye, W.; Zhang, W.; Liu, T.; Huang, Z.; Zhu, M.; Chen, Y.; Li, H.; Li, S. De Novo Transcriptome Sequencing of the Deep-Sea-Derived Fungus Dichotomomyces cejpii and Analysis of Gliotoxin Biosynthesis Genes. Int. J. Mol. Sci. 2018, 19, 1910.
- Ye, W.; Li, S.; Liu, S.; Kong, Y.; Zhang, W.; Liu, S.; Liu, T.; Zhang, W. Characterization of novel gliotoxin biosynthesis-related genes from deep-sea-derived fungus Geosmithia pallida FS140. Biochimie 2021, 191, 1–10.
- Scharf, D.H.; Brakhage, A.A.; Mukherjee, P.K. Gliotoxin–bane or boon? Environ. Microbiol. 2016, 18, 1096–1109.
- Ben-Ami, R.; Lewis, R.E.; Leventakos, K.; Kontoyiannis, D.P. Aspergillus fumigatus inhibits angiogenesis through the production of gliotoxin and other secondary metabolites. Blood 2009, 114, 5393–5399.
- Erjavec, Z.; Kluin-Nelemans, H.; Verweij, P. Trends in invasive fungal infections, with emphasis on invasive aspergillosis. Clin. Microbiol. Infect. 2009, 15, 625–633.
- Ben-Ami, R.; Lewis, R.E.; Kontoyiannis, D.P. Enemy of the (immunosuppressed) state: An update on the pathogenesis of Aspergillus fumigatus infection. Br. J. Haematol. 2010, 150, 406–417.
- Lee, H.J.; Lee, J.H.; Hwang, B.Y.; Kim, H.S.; Lee, J.J. Anti-angiogenic activities of gliotoxin and its methylthio-derivative, fungal metabolites. Arch. Pharm. Res. 2001, 24, 397–401.
- Ben-Ami, R. Angiogenesis at the mold–host interface: A potential key to understanding and treating invasive aspergillosis. Future Microbiol. 2013, 8, 1453–1462.
- Dikmen, M.; Cantürk, Z.; Engur, S.; Kaya Tilki, E. Inhibitory effects of secondary metabolites of halotolerant Pythium ultimum terreus on angiogenesis. Biomed. Res. 2017, 28, 8.
- Maulik, N. Redox signaling of angiogenesis. Antioxid. Redox Signal. 2002, 4, 805–815.
- Orciuolo, E.; Stanzani, M.; Canestraro, M.; Galimberti, S.; Carulli, G.; Lewis, R.; Petrini, M.; Komanduri, K.V. Effects of As-pergillus fumigatus gliotoxin and methylprednisolone on human neutrophils: Implications for the pathogenesis of invasive as-pergillosis. J. Leukoc. Biol. 2007, 82, 839–848.
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 775–787.
- Lignelli, E.; Palumbo, F.; Myti, D.; Morty, R.E. Recent advances in our understanding of the mechanisms of lung alveolarization and bronchopulmonary dysplasia. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L832–L887.
- Ben-Ami, R.; Albert, N.D.; Lewis, R.E.; Kontoyiannis, D.P. Proangiogenic Growth Factors Potentiate In Situ Angiogenesis and Enhance Antifungal Drug Activity in Murine Invasive Aspergillosis. J. Infect. Dis. 2013, 207, 1066–1074.
- Bondy, G.S.; Pestka, J.J. Immunomodulation by fungal toxins. J. Toxicol. Environ. Health Part B Crit. Rev. 2000, 3, 109–143.
- Higurashi, H.; Arai, M.; Watanabe, A.; Igari, H.; Seki, N.; Kamei, K.; Kuriyama, T. Gene expression profiling of polymorphonuclear leukocytes treated with the culture filtrate of Aspergillus fumigatus and gliotoxin. Microbiol. Immunol. 2007, 51, 407–419.
- König, S.; Pace, S.; Pein, H.; Heinekamp, T.; Kramer, J.; Romp, E.; Straßburger, M.; Troisi, F.; Proschak, A.; Dworschak, J.; et al. Gliotoxin from Aspergillus fumigatus Abrogates Leukotriene B4 Formation through Inhibition of Leukotriene A4 Hydrolase. Cell Chem. Biol. 2019, 26, 524–534.
- Coméra, C.; André, K.; Laffitte, J.; Collet, X.; Galtier, P.; Maridonneau-Parini, I. Gliotoxin from Aspergillus fumigatus affects phagocytosis and the organization of the actin cytoskeleton by distinct signaling pathways in human neutrophils. Microbes Infect. 2007, 9, 47–54.
- Speth, C.; Kupfahl, C.; Pfaller, K.; Hagleitner, M.; Deutinger, M.; Würzner, R.; Mohsenipour, I.; Lass-Flörl, C.; Rambach, G. Gliotoxin as putative virulence factor and immunotherapeutic target in a cell culture model of cerebral aspergillosis. Mol. Immunol. 2011, 48, 2122–2129.
- Kroll, M.; Arenzana-Seisdedos, F.; Bachelerie, F.; Thomas, D.; Friguet, B.; Conconi, M. The secondary fungal metabolite gliotoxin targets proteolytic activities of the proteasome. Chem. Biol. 1999, 6, 689–698.
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-κB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 2001, 1, 287–296.
- Dev, A.; Iyer, S.; Razani, B.; Cheng, G. NF-κB and innate immunity. Curr. Top. Microbiol. Immunol. 2011, 349, 115–143.
- Bacher, S.; Schmitz, M.L. The NF-κB pathway as a potential target for autoimmune disease therapy. Curr. Pharm. Des. 2004, 10, 2827–2837.
- Kupfahl, C.; Heinekamp, T.; Geginat, G.; Ruppert, T.; Härtl, A.; Hof, H.; Brakhage, A.A. Deletion of the gliP gene of Aspergillus fumigatus results in loss of gliotoxin production but has no effect on virulence of the fungus in a low-dose mouse infection model. Mol. Microbiol. 2006, 62, 292–302.
- Schlam, D.; Canton, J.; Carreño, M.; Kopinski, H.; Freeman, S.A.; Grinstein, S.; Fairn, G.D. Gliotoxin suppresses macrophage immune function by subverting phosphatidylinositol 3, 4, 5-trisphosphate homeostasis. MBio 2016, 7, e02242–15.
- Haun, F.; Neumann, S.; Peintner, L.; Wieland, K.; Habicht, J.; Schwan, C.; Østevold, K.; Koczorowska, M.M.; Biniossek, M.; Kist, M.; et al. Identification of a novel anoikis signalling pathway using the fungal virulence factor gliotoxin. Nat. Commun. 2018, 9, 3524.
- Jia, X.; Chen, F.; Pan, W.; Yu, R.; Tian, S.; Han, G.; Fang, H.; Wang, S.; Zhao, J.; Li, X.; et al. Gliotoxin promotes Aspergillus fumigatus internalization into type II human pneumocyte A549 cells by inducing host phospholipase D activation. Microbes Infect. 2014, 16, 491–501.
- Li, X.; Gao, M.; Han, X.; Tao, S.; Zheng, D.; Cheng, Y.; Yu, R.; Han, G.; Schmidt, M.; Han, L. Disruption of the Phospholipase D Gene Attenuates the Virulence of Aspergillus fumigatus. Infect. Immun. 2011, 80, 429–440.
- Gayathri, L.; AkbκBarsha, M.A.; Ruckmani, K. In vitro study on aspects of molecular mechanisms underlying invasive asper-gillosis caused by gliotoxin and fumagillin, alone and in combination. Sci. Rep. 2020, 10, 14473.
- Fraga-Silva, T.F.D.C.; Mimura, L.A.N.; Leite, L.D.C.T.; Borim, P.A.; Ishikawa, L.L.W.; Venturini, J.; De Arruda, M.S.P.; Sartori, A. Gliotoxin Aggravates Experimental Autoimmune Encephalomyelitis by Triggering Neuroinflammation. Toxins 2019, 11, 443.
- Herfarth, H.; Brand, K.; Rath, H.; Rogler, G.; Schölmerich, J.; Falk, W. Nuclear factor-κB activity and intestinal inflammation in dextran sulphate sodium (DSS)-induced colitis in mice is suppressed by gliotoxin. Clin. Exp. Immunol. 2000, 120, 59–65.
- Oh, J.; Hur, J.; Kim, Y.; Kwon, Y.-M.; Kim, K.; Chung, Y.; Choi, M. Gliotoxin Protects Trinitrobenzene Sulfonic Acid-Induced Colonic Damage through Induction of Heme Oxygenase-1. Toxicol. Res. 2004, 20, 293–298.
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119.
- Waring, P.; Sjaarda, A.; Lin, Q.H. Gliotoxin inactivates alcohol dehydrogenase by either covalent modification or free radical damage mediated by redox cycling. Biochem. Pharmacol. 1995, 49, 1195–1201.
- Yoshida, L.S.; Abe, S.; Tsunawaki, S. Fungal Gliotoxin Targets the Onset of Superoxide-Generating NADPH Oxidase of Human Neutrophils. Biochem. Biophys. Res. Commun. 2000, 268, 716–723.
- Konstantinovas, C.; de Oliveira Mendes, T.A.; Vannier-Santos, M.A.; Lima-Santos, J. Modulation of human immune response by fungal biocontrol agents. Front Microbiol. 2017, 8, 39.
- Bernardo, P.H.; Brasch, N.; Chai, C.L.; Waring, P. A Novel Redox Mechanism for the Glutathione-dependent Reversible Uptake of a Fungal Toxin in Cells. J. Biol. Chem. 2003, 278, 46549–46555.
- Choi, H.S.; Shim, J.S.; Kim, J.-A.; Kang, S.W.; Kwon, H.J. Discovery of gliotoxin as a new small molecule targeting thioredoxin redox system. Biochem. Biophys. Res. Commun. 2007, 359, 523–528.
- Piaz, F.; Braca, A.; Belisario, M.; De Tommasi, N. Thioredoxin System Modulation by Plant and Fungal Secondary Metabolites. Curr. Med. Chem. 2010, 17, 479–494.
- Tsunawaki, S.; Yoshida, L.S.; Nishida, S.; Kobayashi, T.; Shimoyama, T. Fungal Metabolite Gliotoxin Inhibits Assembly of the Human Respiratory Burst NADPH Oxidase. Infect. Immun. 2004, 72, 3373–3382.
- Warris, A.; Ballou, E.R. Oxidative responses and fungal infection biology. Semin. Cell Dev. Biol. 2019, 89, 34–46.
- Nishida, S.; Yoshida, L.S.; Shimoyama, T.; Nunoi, H.; Kobayashi, T.; Tsunawaki, S. Fungal Metabolite Gliotoxin Targets Flavocytochrome b558 in the Activation of the Human Neutrophil NADPH Oxidase. Infect. Immun. 2005, 73, 235–244.
- Li, G.; Kobayashi, T.; Tsunawaki, S.; Ogawa, Y.; Seguchi, H. Gliotoxin Inhibits Superoxide Production and Exocytosis of the Oxidant-producing Intracellular Compartments in Human Neutrophils Stimulated with Phorbol Myristate Acetate. Acta Histochem. Cytochem. 2004, 37, 183–189.
- Zhang, B.; Zhang, J.; Peng, S.; Liu, R.; Li, X.; Hou, Y.; Fang, J. Thioredoxin reductase inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 547–556.
- Kweon, Y.-O.; Paik, Y.-H.; Schnabl, B.; Qian, T.; Lemasters, J.J.; Brenner, D.A. Gliotoxin-mediated apoptosis of activated human hepatic stellate cells. J. Hepatol. 2003, 39, 38–46.
- Miralles, L.M. Investigating and Exploiting Fungal Natural Products in Aspergillus Spp.; National University of Ireland: Maynooth, Ireland, 2015.
- Chai, C.; Waring, P. Redox sensitive epidithiodioxopiperazines in biological mechanisms of toxicity. Redox Rep. 2000, 5, 257–264.
- Nouri, M.A.; Al-Halbosiy, M.M.; Dheeb, B.I.; Hashim, A.J. Cytotoxicity and genotoxicity of gliotoxin on human lymphocytes in vitro. J. King Saud Univ. Sci. 2015, 27, 193–197.
- Nieminen, S.M.; Mäki-Paakkanen, J.; Hirvonen, M.R.; Roponen, M.; von Wright, A. Genotoxicity of gliotoxin, a secondary metabolite of Aspergillus fumigatus, in a battery of short-term test systems. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2002, 520, 161–170.
- Golden, M.C.; Hahm, S.J.; Elessar, R.; Saksonov, S.; Steinberg, J. DNA damage by gliotoxin from Aspergillus fumigatus. An occupational and environmental propagule: Adduct detection as measured by 32P DNA radiolabelling and two-dimensional thin-layer chromatography: DNA-Schädigung durch Gliotoxin von Aspergillus fumigatus. Eine arbeits-und umwelt-medizinische Studie: Addukt-Nachweis mittels 32P-Markierung und zweidimensionaler Dünnschichtchromatographie. Mycoses 1998, 41, 97–104.
- Suen, Y.K.; Fung, K.P.; Lee, C.Y.; Kong, S.K. Gliotoxin induces apoptosis in cultured macrophages via production of reactive oxygen species and cytochrome c release without mitochondrial depolarization. Free Radic. Res. 2001, 35, 1–10.
- Eichner, R.D.; Waring, P.; Geue, A.M.; Braithwaite, A.W.; Müllbacher, A. Gliotoxin causes oxidative damage to plasmid and cellular DNA. J. Biol. Chem. 1988, 263, 3772–3777.
- Lin, H.; Wang, Q.; Niu, Y.; Gu, L.; Hu, L.; Li, C.; Zhao, G. Antifungal and anti-inflammatory effect of punicalagin on murine Aspergillus fumigatus keratitis. Curr. Eye Res. 2021.
- Ramalingam, S.; Bahuguna, A.; Kim, M. The effects of mycotoxin patulin on cells and cellular components. Trends Food Sci. Technol. 2018, 83, 99–113.

