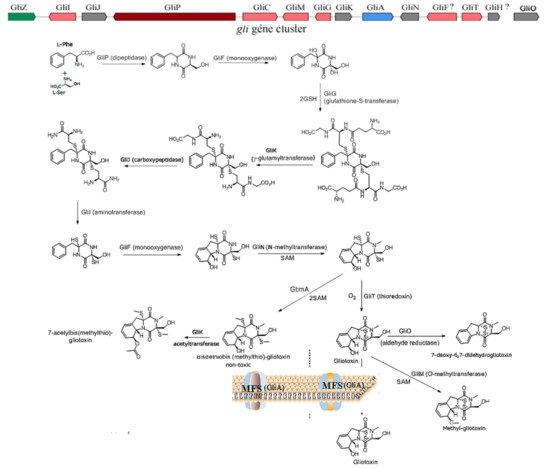
2.1. The Inhibition of Angiogenesis
Previous studies have demonstrated that gliotoxin can inhibit angiogenesis in humans with the symptom of invasive aspergillosis, which was intermediated by the gliotoxin produced by
A. fumigatus [12][22]. Meanwhile, invasive aspergillosis is the most common infected fungal disease that causes high mortality, especially for immune deficiency patients, because of the deficiency in the approaches for diagnosis and treatment
[13][23]. Additionally, the incidence rate of invasive aspergillus is increasing year by year due to the wider application of organ transplants and antibiotics
[13][23]. As a consequence, it is important to elucidate the mechanism of aspergillosis mediated by gliotoxin. Gliotoxin can repress angiogenesis in the lung of mice and patients suffering from aspergillosis; furthermore,
A. fumigatus could impede capillary tube formation in a murine model of cutaneous infection via the generation of secondary metabolites, especially gliotoxins
[14][24]. Additionally, pure gliotoxin can also suppress angiogenesis and endothelial cell migration in vitro
[12][15][22,25]. The deletion of
gliP in the biosynthetic gene cluster and LaeA global transcriptional regulator can significantly attenuate the angiogenic effect caused by
A. fumigatus [12][16][22,26], thus further supporting the function of angiogenesis caused by gliotoxin
[17][27]. In addition, the immune response can also be inhibited via the antiangiogenic effect mediated by gliotoxin, thus further deteriorating the symptom of invasive aspergillus.
The proangiogenic and antianogentic effects caused by
A. fumigatus has been investigated. The antiangiogenesis effect of gliotoxin produced by
A. fumigatus was exerted via the reduction of the generation of ROS and, thereby, the suppression of NF-κB
[18][28]. Furthermore, gliotoxin can also repress the proliferation of polymorphonuclear leucocytes
[19][29]. Moreover, gliotoxin can suppress the generation of NF-κB, thus blocking the production of cytokines and inflammatory factors to reduce the angiogenic effect
[20][30]. To compensate the antigiogenic effect, thus protecting the host, the infection with
A. fumigatus triggers proangiogenic effects, including the production of proinflammatory factors such as tumor necrosis factor alpha (TNFα), hypoxia-inducible factor 1-alpha (HIF1α), and interleukin (IL-8), thus recruiting the polymorphonuclear leucocytes (PMNLs) and leading to the burst of ROS, including H
2O
2. The gene ratio of ROS can induce the upregulation of NF-κB, thus elevating the expression of vascular endothelial growth factor and other proangiogenic factors to compensate the antanginogenic effect caused by
A. fumigatus [12][21][22,31]. Moreover, gliotoxin can abrogate the angiogensis induced by H
2O
2 in low concentrations, and gliotoxin can completely inhibit the angiogenic effect in immunosuppressive mice treated with cyclophosphamide via the abolishment of the production of ROS, which is mediated by gliotoxin-catalyzed reduction by the assistance of NAPDH
[22][32]. Therefore, these studies also provide strategies for the prevention of angiogenic effects of invasive aspergillus via the upregulation of proangiogenic mediators, including NF-κB, cytokines, and vascular endothelial growth factor (VGEFs). Different concentrations of gliotoxins can induce different proangiogenic and antiangiogenic effects; thus, the angiogenic effect can be balanced via the control of gliotoxin concentrations.
2.2. Immunosuppressive Activity
Gliotoxin has been reported to induce immunosuppressive activity via cell apoptosis mediated by the repression of phagocytosis and macrophages
[23][33] as well as the suppression of the proliferation of giant cells and T-cells
[23][33]. Moreover, gliotoxin can further deteriorate the diseases caused by fungi infection via the inhibition of the activities of immune-related cells
[23][24][33,34]. The amounts of immune cells, including leucocyte, lymphocyte, and Langerhans cells, significantly decreased after the treatment with gliotoxins in adult camels for 7 days. Furthermore, gliotoxin from
A. fumigatus abolish the generation of oleukotriene B4 through the suppression of leukotriene A4 hydrolase, thus exerting the function of immunosuppression
[25][35]. In addition, the knockout of
gliP and
gliZ genes attenuated the toxicity of gliotoxin towards nonneutrophil cells in mice
[26][36]; 50 nM of gliotoxin can exert immunosuppressive activity via the suppression of the phagocytosis of astrocytes
[14][27][24,37], which led us to exploit low-dose gliotoxins as immunosuppressive agents during an organ-transplant operation, thus reducing immunological rejection in organ transplant patients.
The immunosuppressive mechanism of gliotoxin has been widely investigated. It is well known that gliotoxin can inhibit the activation of NF-κB mediated by the repression of IκBα degradation caused by the chymotrypsin-like activity of the 20S proteasome
[28][38]. The disulfide of glitoxin can target the protease, which can be reversed by reducing agent dithiothreitol, which can be reduced from gliotoxin to dithiol gliotoxin. Low doses of gliotoxin, with a concentration of 1 μM, exhibited 63% inhibition of the LLVY-amc peptide-hydrolyzing activity mediated by the 20S proteasome, suggesting the high immunosuppressive efficiency of gliotoxin
[28][38].
NF-κB is a kind of inducible transcriptional factor; some examples are NF-κB1 (p50), NF-κB2 (p52), RelA, RelB, and c-Rel. A typical active NF-κB is the P50-P65 dimer that can bind DNA; thus, activating NF-κB will modulate inflammation and innate and adaptive immune system reactions. The IκBα protein can inhibit the activity of NF-κB in the cytoplasm via binding with NF-κB, which can be activated by the degradation of the IκBα protein, mediated by the 20S protease, and by the binding of the DNA in the nucleus
[29][30][31][39,40,41]. Glitoxin can stabilize the expression level of IκBα via the inhibition of the 20S protease, blocking the activation of NF-κB and thus exert the function of immunosuppression; the disulfide bond in gliotoxin is essential for its immunorepression activity
[28][38]. In consequence, the disruption of
gliP can abolish the immunosuppression function of
A. fumigatus via the suppression of gliotoxin production
[32][42]. Furthermore, gliotoxin can assist
A. fumigatus in achieving immune escape from the phagocytosis of macrophages by targeting the metabolism of phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3]
[33][43]; the gliotoxin can promote the immune escape of
A. fumigatus by decreasing the content of intracellular lipid acyl inositol 3,4,5-trisphosphate
[33][43], thus leading to the function deficiency of the integrin and actin backbone and preventing cell membrane protrusion extension
[34][44], further impairing the function of macrophages. Furthermore, it was reported that gliotoxin contributed to the invasion of
A. fumigatus spores into cells via the activation of phospholipase D in lung epithelial cells and by inducing actin backbone rearrangement
[35][36][45,46], but the specific regulatory mechanism remains obscure.
2.3. Inflammatory and Anti-Inflammatory Effects
Gliotoxin can also initiate the production of inflammatory factors, thus eliciting further cytotoxicity. Gliotoxin incubation with A459 cells can induce the secretion of proinflammatory cytokines, including IL-4, IL-8, and IL-10, whereas the upregulation of IL-6 and IL-8 cytokines was observed in gliotoxin-treated L132 cells, with only IL-8 upregulation observed in A459 and L132 cells treated with fumagillin
[24][37][34,47]. In addition, the expression of a few cytokines, such as IL-1β, IL-17, interferon-gamma (IFN-γ), TNF-α, and granulocyte macrophage-colony stimulating factor (GM-CSF), was downregulated in the two cells treated with gliotoxin and fumagillin, suggesting neutrophil-mediated tissue damage after
A. fumigutas infection
[37][47]. The results also suggest a stronger inflammation-eliciting effect by gliotoxin compared with fumagillin. Gliotoxin was administrated to female C57BL/6 mice immunized with myelin oligodendrocyte glycoprotein with a dose of 1 mg/kg, which can aggravate the symptom of neuroinflammation via the upregulation of inflammatory genes, including T-box transcription factor (TBX21), inducible nitric oxide synthase (iNOS), arginase 1 (ARG1), and cytokines such as IFN-γ, IL-17, and IL-2
[38][48].
In contrast, in vivo anti-inflammatory activity was also investigated. Herfarth et al. reported that gliotoxin can suppress intestinal inflammation in dextran sulfate sodium (DSS)-induced colitis mediated by NF-κB activation
[39][49]. The expression of cytokines, including TNF-α, was downregulated in RAW-264.7 mouse macrophage-like cells; meanwhile, TNF-α and IL-1α mRNA expressions were also significantly suppressed in DSS-induced colitis mice by gliotoxin treatment for 8 days, thus leading to the alleviation of the symptom of colonic inflammation in mice
[39][49]. Furthermore, the gliotoxin treatment also reduced the DNA-binding activity of NF-κB, thus impeding the inflammation mediated by NF-κB activation in the colon
[20][30]. In addition, gliotoxin can ameliorate trinitrobenzene sulfonic acid (TNBS)-induced mouse colitis via the suppression of the expression levels of TNF-, IL-1, and intercellular adhesion molecule-1 (ICAM-1) proteins as well as the upregulation of heme oxygenase-1, thus indicating the potential application of gliotoxins in the clinical treatment of Crohn’s disease
[40][50] via the induction of hemooxygenase-1, which can oxidize low-density lipoproteins with the potential to form chemo-attractants, thus alleviating the inflammation
[41][51].
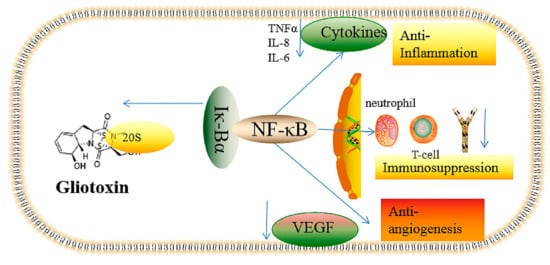
In summary, gliotoxin’s toxic effects, including the antiangiogenesis and inflammatory-related effects mediated by NF-κB activity and redox cycling, are summarized in Figure 1. Gliotoxin can target the 20S proteasome, which is responsible for the degradation of the IK-Bα protein that specifically binds to NF-κB; thus, gliotoxin can suppress NF-κB activity. NF-κB is closely associated with the function of angiogenesis, immune response, and cell proliferation; thus, gliotoxin treatment can induce the effects of immunosuppression, antiangiogenesis, and anti-inflammation (Figure 2).
Figure 2. The cytotoxic effects, including immunosuppression, antiangiogenesis, and anti-inflammation, of gliotoxin mediated by NF-κB. The arrow to the left indicates that gliotoxin can detach 20S protease from Iκ-Bα, thus inhibiting the activation of NF-κB. The downward arrow indicates the down-regulation caused by the inactivation of NF-κB mediated by gliotoxin.
2.4. Inducing the Production of ROS and the Inhibition of Peroxidase
Gliotoxin can induce the production of reactive oxygen species (ROS) via intracellular redox cycling
[42][52], thus resulting in damage from the peroxidation of macromolecules and biofilms, mediated by oxidative stress and the abrogation of antioxidation. In human neutrophils, reduced gliotoxin acts as a generator of superoxide by inhibiting NADPH oxidase
[43][53]. Alternatively, gliotoxin can also served as an antioxidant. Reduced nicotinamide adenine dinucleotid phosphate (NADPH) peroxidase can only produce low contents of ROS to scavenge pathogenic bacteria and fungi, thus protecting the hosts. Meanwhile, Tsunawaki et al. reported that gliotoxin can inhibit the enzymatic activity of NADPH peroxidase with a concentration of higher than 1 μg/mL, thus causing damage to the host, whereas the NADPH peroxidase in neutrophils was not inhibited by the treatment with this dose of gliotoxin. Thus, different doses of gliotoxin induce different oxidative damage and different physiological effects; 30–100 ng/mL gliotoxin can inhibit the phagocytosis of zymosan without affecting ROS production. Gliotoxin can also lead to the reshaping of the actin cytoskeleton
[44][54], thus contributing to cell shrinkage and filopodia disappearance, which can be reversed by Cyclic Adenosine monophosphate (cAMP) antagonist Rp-cAMP; however, gliotoxin-induced phagocytosis can not be inversed by cAMP
[44][54].
The detailed mechanism of the gliotoxin-induced mechanism mediated by ROS has been investigated. Bernardo et al. described a redox-uptake system mediated by gliotoxin
[45][55]: intracellular gliotoxin can be reduced to dithiol gliotoxin by a cellular reducing agent, thus releasing electrons to cellular O
2 to form O
2-mediated gliotoxin, which can yield ROS. Furthermore, dithiol gliotoxin can also be oxidized into gliotoxin by cellular O
2 in turn, thereby resulting in the accumulation of ROS via the redox-uptake system mediated by gliotoxin. Thus, gliotoxin can cause cell peroxide damage mediated by the redox-uptake system in a dose-dependent manner.
On the other hand, gliotoxin can also be explored as an antioxidant. Gliotoxins exhibited the ability to reduce H
2O
2 to H
2O in a thioredoxin redox system composed of thioredoxin, thioredoxin reductase, and NADPH
[46][47][56,57], whereas gliotoxin did not show H
2O
2-reducing activity in a glutathione (GSH) / glutathione disulfide (GSSG) reductase system. The H
2O
2-reducing activity in the yeast Trx system depends on the concentrations of gliotoxin and H
2O
2, which was applicable to the rat Trx system
[47][57], suggesting gliotoxin as a potent antioxidant for mammalian cells. The in vitro H
2O
2-reducing activity of gliotoxin in Hela cells was also investigated; the results indicated that the H
2O
2 level was significantly decreased by the addition of gliotoxin, with concentrations ranging from 40–160 nmol, in a dose-dependent manner in Hela cells, which was demonstrated by 2′,7′-dichlorofluorescein diacetate (DCFH-DA) probes
[47][57]. In addition, gliotoxin can target NADPH oxireductase to prevent the onset of O
2- generation in human neutrophils
[48][58] and lead to the release of cytochrome c through a reaction with intracellular reductants in an oxygen-dependent manner; the prevention of redox cycling facilitated the damage of gliotoxin to NADPH oxireductase with IC
50 of 9 nM
[49][59]. Furthermore, the detailed targeting of gliotoxin to the NADPH oxidase system was investigated by a cell-free activation assay with NADPH oxidase components. The ROS generation ability of the membrane was reduced by 40% after gliotoxin treatment
[19][49][50][29,59,60], and the in vitro addition of gliotoxin to flavocytochrome b558 and cytosolic components also exhibited an inhibition rate of 50% with the concentration of 3.3 μM
[50][60], suggesting that flavocytochrome b558 is the target of gliotoxin. Moreover, the cell surface of alkaline phosphatase activity, which is a marker enzyme of oxidant-producing intracellular compartments (OPICs), is also suppressed by gliotoxin in human neutrophils stimulated with phorbol myristate acetate (PMA) as well as the repression of NADPH oxidase
[51][61], thus contributing to the inhibition of superoxide production in PMA-treated human neutrophils.
Moreover, the block of oxidative stress by gliotoxin would impede the proliferation of human umbilical vein endothelial cells in a dose-dependent manner with the IC
50 of gliotoxin of 250 nM
[46][52][56,62], which was attributed to the fact that oxidative stress can trigger angiogenesis. Nevertheless, gliotoxin exhibited cytotoxicity to human umbilical vein endothelial cells (HUVECs) with concentrations higher than 500 nM
[46][52][56,62], whereas there was no obvious cytotoxicity with concentrations lower than 300 nM, which is consistent with a previous study that found that gliotoxin induces apoptosis in activated human hepatic stellate cells with concentrations ranged from 0.3 to 7.5 μM
[53][63]. To further elucidate the role of H
2O
2 and gliotoxin in angiogenesis, 100 nM H
2O
2 was added to induce the sprout of endothelial cells and the formation of thick tubes, which were inhibited by the addition of gliotoxin with concentrations of 125 to 250 nM
[52][62]. In addition, gliotoxin potently inhibited the H
2O
2-stimulated invasion of HUVECs to the control level at 125 nM
[52][62]. Thus, gliotoxin can exert the function of antiangiogenesis via the regulation of the intracellular redox state. The thioredoxin-SH group can reduce the gliotoxin to dithiol gliotoxin while leading to the reduction of H
2O
2 to H
2O
[46][54][55][56,64,65], thereby alleviating the angiogenic and invasive effects mediated by H
2O
2. Therefore, low doses of gliotoxin show great potential to be developed as an antioxidant to encounter the peroxide damage caused by H
2O
2.
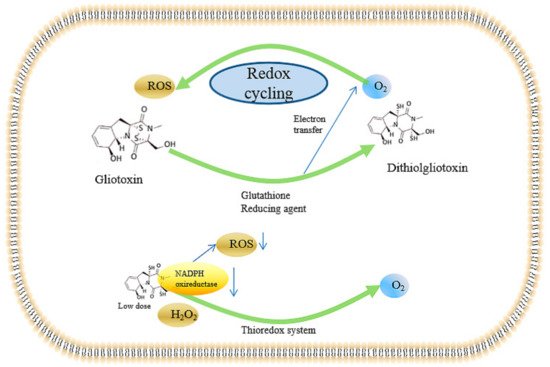
Taken together, the ROS-inducing effects mediated by the redox cycling system caused by gliotoxin are summarized in Figure 3. Gliotoxin can be reduced to dithiol gliotoxin by in vivo reducing agents, including dithiothreitol (DTT), glutathione, and reducing enzymes, thus providing electrons to oxygen atoms and, thereby, producing intracellular ROS. Excessive ROS can cause peroxide damage and single- and double-strand breaks, causing great damage to the human body. In contrast, gliotoxin can be served as an antioxidant in the presence of the thioredoxin redox system through the inhibition of NADPH oxidase. Moreover, both gliotoxin and ditholgliotoxin are cytotoxic; however, gliotoxin is more toxic, probably due to the existence of the disulfide bridge, which can bind to many physiological-related proteins.
Figure 3. Gliotoxin-mediated redox cycling system and the antioxidant effect. The downward arrow indicates the ROS content was decreased by low dose of gliotoxin via the inhibition of NADPH oxireducatase activity in the presence of thioredox system.
2.5. Genotoxicity of Gliotoxin
Gliotoxin can also cause genotoxicity mediated by DNA damage in vitro and in vivo. Ditholgliotoxin can cause single- and double-stranded breaks in the presence of DNA and Fe
3+, which was determined by neutral agarose gel electrophoresis
[56][57][66,67]. Gliotoxin did not induce single- and double-stranded breaks except by the addition of reducing agents, including glutathione, DTT, and pyridine reducing enzymes
[57][67], suggesting the genotoxicity was probably mediated by ditholgliotoxin and this DNA damage effect can be abrogated by metal chelator and catalase. This supports that the DNA damage effect was mediated by the ROS produced during the redox cycling process to form hydroxylated and other altered DNA products, which was determined by 32P DNA radiolabelling and two-dimensional thin-layer chromatography
[58][68].
DNA damage was observed in mouse RAW264.7 macrophages after gliotoxin treatment for 2 h in plain medium in a single cell electrophoresis assay
[57][59][60][61][67,69,70,71]; however, a clear, dose-related increase in sister-chromatid exchange was not observed in Chinese hamster ovary (CHO) cells
[62][72]. Gliotoxin can also cause mitochondrial damage, thus producing ROS to induce DNA damage and causing toxicity to lung epithelial cells A459 and L132; this is stronger than another toxin fumagillin produced by
A. fumigatus [37][47]. It is also speculated that gliotoxin can induce lung epithelial cells damage mediated by endoplasmic reticulum (ER) stress due to the observation of the accumulation of unfolded proteins in the lumen of the ER. Additionally, the cytotoxicity of gliotoxin towards different organ-derived cells is as follows: renal epithelial cells > type II epithelial cells > hepatocytes > normal lung epithelial cells
[37][47], indicating that gliotoxin has selective cytotoxicity towards organ-derived cells to some extent.