Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rosaria Saletti | + 3752 word(s) | 3752 | 2021-12-01 07:34:34 | | | |
| 2 | Camila Xu | -44 word(s) | 3708 | 2021-12-23 02:02:36 | | | | |
| 3 | Camila Xu | -44 word(s) | 3708 | 2021-12-23 02:07:37 | | | | |
| 4 | Camila Xu | -44 word(s) | 3708 | 2021-12-23 02:08:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Saletti, R. Voltage-Dependent Anion Selective Channel. Encyclopedia. Available online: https://encyclopedia.pub/entry/17413 (accessed on 26 July 2026).
Saletti R. Voltage-Dependent Anion Selective Channel. Encyclopedia. Available at: https://encyclopedia.pub/entry/17413. Accessed July 26, 2026.
Saletti, Rosaria. "Voltage-Dependent Anion Selective Channel" Encyclopedia, https://encyclopedia.pub/entry/17413 (accessed July 26, 2026).
Saletti, R. (2021, December 21). Voltage-Dependent Anion Selective Channel. In Encyclopedia. https://encyclopedia.pub/entry/17413
Saletti, Rosaria. "Voltage-Dependent Anion Selective Channel." Encyclopedia. Web. 21 December, 2021.
Copy Citation
VDAC (voltage-dependent anion selective channel) proteins, also known as mitochondrial porins, are the most abundant proteins of the outer mitochondrial membrane (OMM) where they play a vital role in various cellular processes, in the regulation of metabolism, and in survival pathways.
voltage dependent anion channel
cysteine overoxidation
deamidation
1. Introduction
1.1. VDAC Isoforms: A Family of Hub Proteins
VDAC (voltage-dependent anion selective channel) proteins, also known as mitochondrial porins, are the most abundant proteins of the outer mitochondrial membrane (OMM) where they play a vital role in various cellular processes, in the regulation of metabolism, and in survival pathways. They mediate the ions and metabolites exchange between mitochondria and the rest of the cell, ensuring good functionality of mitochondrial complexes and energy production [1].
In higher eukaryotes, there are three VDAC isoforms (VDAC1, VDAC2, VDAC3) encoded by separate genes located on different chromosomes [2]. These pore-forming proteins have a similar molecular weight (30 kDa) and highly conserved sequences of about 280 amino acids with the exception of VDAC2 which has the N-terminal moiety of 11 residues longer than the other isoforms.
The evolutionary analysis indicates VDAC3 as the oldest isoform, while VDAC1 is considered the youngest mitochondrial porin [3][4].
The experimental 3D structures of mouse and human VDAC1 isoform have been determined using X-ray crystallography and NMR [5][6][7]. These analyses revealed a structure constituted by 19 β-strands arranged to form a trans-membrane β-barrel and by a region containing α-helix at the N-terminus of the protein. The barrel is organized as a regular antiparallel array of β-strands with the exception of strands 1 and 19 that run in parallel. The amphipathic α-helix tail is located inside the pore. However, the exact position and local structure of this segment are still elusive since these features are not perfectly overlapping in the available X-ray and NMR structures [5][6][7].
Recently, the structure of zebrafish VDAC2 was solved at a high resolution, confirming the same β-barrel arrangement as VDAC1 [8]. Zebrafish VDAC2 has one cysteine residue in the sequence and lacks the 11 amino acid longer N-terminal sequence present in mammalian VDACs.
The VDAC3 structure has not yet been determined. Several bioinformatic predictions, based on the large sequence similarity, proposed a barrel core such as the other VDAC isoforms [9]. Despite the high sequence similarity and structural homology, VDAC isoforms display different functional properties within the cell.
Analysis of the expression levels of human VDAC isoforms in HeLa cells, determined by real-time PCR, suggests that VDAC1 is the most abundant isoform, ten times more abundant compared to VDAC2 and hundred times more abundant compared to VDAC3, the least characterized of the isoforms. In addition, the overexpression of each single VDAC isoform affects the mRNA levels of the other two isoforms, suggesting that the ratios between VDAC isoforms are subjected to a reciprocal control that avoids an imbalance among these proteins [10].
Although the three isoforms show a common involvement in cellular bioenergetics maintenance, VDAC1 and VDAC2 have specialized functions in programmed cell death. For VDAC3 isoform, recent studies indicate a central role in ROS metabolism and in mitochondrial quality control [11].
The functions of VDACs are several-fold and some of these depend on, or are affected by, interaction with other cytosolic and mitochondrial proteins. Due to their localization at the OMM, VDACs are considered to be hub proteins, interacting with over 200 proteins in order to integrate mitochondrial functions with the rest of the cellular activities [12][13]. Thus, VDAC isoforms appears to be a junction for a variety of signals associated with different pathways related to cell survival or programmed death. Furthermore, the function of VDACs and their interactions with other proteins are affected by post-translational modifications (PTMs) [14]. Unfortunately, PTMs of VDAC proteins represent a little explored field, mainly because discovery and characterization of PTM in these proteins is very challenging, due to their poor solubility and impossibility to isolate single isoforms. Only in recent years has the increasing number of tools aimed at identifying and quantifying PTMs increased, improving the knowledge in this field and in the mechanisms that regulate functions and interactions of mitochondrial porins. In particular, the development of nano-reversed phase ultra-high-performance liquid chromatography (nanoRP-UHPLC) and ultra-sensitive high-resolution mass spectrometry (HRMS) methods has played a key role in this field. The findings obtained on VDAC PTMs using such methodologies, which have permitted an in-depth characterization of these very hydrophobic trans-membrane pore proteins, are summarized in this review.
1.2. VDACs as Main Players in Mediating and Regulating Mitochondrial Functions with Cellular Activities
The location in the OMM allows the VDAC proteins to act as anchor points for diverse sets of molecules that interact with mitochondria. In this way, VDACs are able to mediate and regulate the integration of mitochondrial functions with cellular activities.
The VDAC interactome includes proteins located in OMM, inner mitochondrial membrane (IMM), intermembrane space (IMS), cytosol, endoplasmic reticulum, plasma membrane and nucleus that are involved in metabolism, apoptosis, signal transduction, protection against ROS, binding to RNA or DNA, and more.
Mobility of VDAC N-terminal α-helix region is important for channel gating but also for interactions with both pro-apoptotic and anti-apoptotic proteins such as Bax, Bak, and Bcl-xL [15][16][17]. VDAC1 is involved in the release of apoptotic factors located in the intermembrane space due to its ability of oligomerizing in dimers, hexamers, and higher-order structures, to form a large pore that allows the passage of cytochrome c and apoptosis inducing factor (AIF) to the cytosol and consequently the activation of programmed cell death. Instead, VDAC2 functions as anti-apoptotic factor and it is upregulated in several debilitating diseases including Alzheimer’s and cancer [18]. This property is probably due to the unique ability of VDAC2 to sequester the pro-apoptotic protein Bak in the OMM and maintain it in the inactive state [11].
VDAC1 displays binding sites, located in its cytosolic moiety, for many metabolic enzymes, such as glyceraldehyde 3-phosphate dehydrogenase, creatine kinase, glycerol kinase, glucokinase, c-Raf kinase, and hexokinase isoforms (I and II), which need preferential access to mitochondrial ATP [19].
Hexokinase interacts through its hydrophobic N-terminal sequence with Glu73 of VDAC1, a binding site localized on one side of the barrel wall, buried in the hydrophobic environment of OMM [20].
It has been demonstrated that treatment of mitochondria with dicyclohexylcarbodiimide (DCCD) inhibits hexokinase–VDAC interaction due to selective chemical modification of Glu73 [21].
Glu73 residue is also the binding site for ceramides, tumor suppressor lipids able to act directly on mitochondria to trigger apoptotic cell death. It is interesting that both VDAC1 and 2 own, in a similar position, a cysteine residue (Cys127 in human VDAC1 and Cys138 in human VDAC2) in the form of sulfonic acid with a strong negative charge resembling that of the glutamate acid residue [22]. Instead, VDAC3 isoform does not show any residue homologous to Cys127/138 or Glu73 embedded in the hydrophobic moiety of the OMM.
VDAC1 presents a cholesterol binding pocket formed, in human isoform, by Ile123, Leu144, Tyr146, Ala151, and Val171 residues [23].
Mitochondrial porins form complexes with other proteins, such as the adenine nucleotide translocase (ANT), the translocator protein (TSPO), also known as the peripheral-type benzodiazepine receptor (PBR), mitochondrial HSP70, and several cytoskeletal proteins such as tubulin, actin, dynein light chain, and gelsolin [11].
The translocator protein interacts directly with all VDAC isoforms. In particular, interaction between TSPO and VDAC1 contributes to regulate the efficiency of mitochondrial quality control mechanisms and inhibits mitophagy, preventing ubiquitination of proteins through downregulation of the PINK1/Parkin pathway [24]. The GxxxG motif presents both in VDAC and in TSPO, and is necessary for this interaction [25]. Moreover, VDAC1 and TSPO, in association with StAR (steroidogenic acute regulatory protein), form the transduceosome, a multi-protein complex involved in cholesterol transport. In a former hypothesis, VDAC1 and TSPO in OMM, ANT in IMM, and Cyclophilin D in the mitochondrial matrix were candidates to constitute the permeability transition pore (PTP), a high conductance and non-specific pore that allows mitochondrial swelling and release of apoptogenic proteins. More recently, it was proposed that PTP could be formed by dimers of the ATP synthase complex [26].
Recent studies have focused attention on the role of VDAC proteins in mitochondrial dysfunction typical of many pathological conditions including stroke, cancer, mitochondrial encephalomyopathies, and aging, as well as neurodegenerative disorders [27][28].
Mass spectrometry analysis revealed the association between VDACs and the ubiquitin ligase Parkin. In presence of damaged mitochondria, as in Parkinson’s disease, Parkin is phosphorylated by PINK1 and consequently ubiquitinates proteins that reside on the OMM, targeting the mitochondria for degradation. Parkin is a cytosolic protein but translocates to the mitochondria to participate in mitochondrial quality control mechanisms. VDAC proteins represent a docking site of Parkin on defective mitochondria [29].
For example, in AD post-mortem brains, in neuroblastoma cells and in an AD mouse model, a direct association was demonstrated between VDAC1, specifically its N-terminal region, and hyper-phosphorylated Tau but also with amyloid beta (Aβ), both in its monomeric and oligomeric forms [31]. These interactions can have a dramatic effect on mitochondrial functions in AD neuron because they block the PTP formation, disrupt the transport of mitochondrial proteins and metabolites, and impair gating, conductance, and physiological interactome of VDACs [32].
In Parkinson’s disease, α-synuclein directly interacts with mitochondria, blocks VDAC1, and impairs metabolite fluxes leading, consequently, to an energetic crisis able to compromise cell viability [33].
In ALS, several SOD1 mutants are able to bind VDAC1 [34]. This interaction impairs ATP/ADP exchange, VDAC1 conductance and mitochondrial membrane potential. Recently, the competition between SOD1G93A and HK1 was demonstrated in binding VDAC1, in NSC34 motor-neuron cell lines [35].
In literature, the role of VDAC1 in neurodegeneration is rather well known; however, the involvement of the other two isoforms in these pathways remains poorly defined. This is likely associated with the relative abundance of VDAC1 compared to other isoforms which are more difficult to isolate in pure form.
Recent studies demonstrated that Cytoskeleton-associated protein 4 (CKAP4), a palmitoylated type II transmembrane protein localized to the endoplasmic reticulum (ER), regulates mitochondrial functions through an interaction with VDAC2 at ER-mitochondria contact sites [36].
VDAC2 binds inositol trisphosphate receptors (IP3R) and regulates the release of Ca2+ from the ER. In addition, several other interaction partners have been reported for VDAC2 isoform, which imply its effect in multiple cellular functions. Specifically, VDAC2 has been linked to many cellular proteins, including apoptotic factors as Bak and Bax, StAR, Metaxin2, eNOS (nitric oxide synthesize), GSK3β, tubulin, and Mcl1 [19]. In addition, VDAC2 and RACK1 (receptor of activated protein kinase C1) function as receptors for lymphocystis disease virus (LCDV) and for bursal disease virus in host cells [37].
VDAC2 together with VDAC3 binds Erastin, the activator of ferroptosis, a new pathway that regulates cell death characterized by the iron-dependent accumulation of lipid hydroperoxides. Interaction between VDAC2/3 and Erastin results in degradation of the channels following activation of ubiquitin protein ligase Nedd4 [38].
Finally, the VDAC3 isoform is associated with cytosolic proteins as tubulins and cytoskeletal proteins, stress sensors, chaperones, and proteasome components, redox-mediating enzymes such as protein disulfide isomerase [39].
2. Proteomics of VDAC Isoforms
2.1. Sample Preparation
Sample preparation has a profound effect on the final results of a proteomic workflow. Protein extraction methods and protein separation techniques should provide an unbiased and reliable map representative of all proteins present in a specific sample. The different extraction and fractionation approaches are based on proteins physicochemical and structural characteristics, such as molecular weight, solubility, hydrophobicity, and isoelectric point. A specific protocol has to be optimized for each particular sample, to maximize protein recovery and minimize the possible proteolysis and amino acid modifications. For these reasons, there is no universal extraction protocol and not a unique buffer composition. Regarding the extraction method, the different strategies available need to be compatible with both the amount of the processed material and the subsequent analytical approach (i.e., separation or MS).
The structural characterization of VDACs presents challenging issues due to their very high hydrophobicity, low solubility, and the impossibility to separate them from other mitochondrial proteins of similar hydrophobicity and to easily isolate each single isoform. In fact, isolation of VDACs has been possible exclusively for plant VDAC isoforms by chromatofocusing, thanks to the absence of phosphorylation sites in their structure [40]. Consequently, it is necessary to analyze them as components of a relatively complex mixture.
A bottom-up proteomic approach was used to investigate the VDAC3 from rat liver mitochondria (rVDAC3) [41]. According with a standard procedure [42], mitochondria were extracted and lysed with a buffer containing 3% Triton X-100 at pH 7.0. The VDAC proteins were partially purified by hydroxyapatite (HTP) chromatography, which allows to obtain a VDACs enriched fraction which comprises also other mitochondria hydrophobic proteins. After precipitation with cold acetone, the protein pellet was solubilized in SDS buffer and loaded on a 17% polyacrylamide gel (1D-SDS-PAGE). The bands in the range 30–35 kDa were manually excised from the gel, cut in small pieces, and subjected to reduction with DTT and alkylation by addition of IAA. Finally, the reduced and carboxyamidomethylated proteins were in gel-digested using trypsin and chymotrypsin, and the resulting peptide mixtures were analyzed by nUHPLC/HRMS [41]. MS data showed that rVDAC3 was found in the whole range 30–35 kDa, together with other proteins, mainly VDAC1, VDAC2, and several other mitochondrial proteins. The reason for VDAC3 electrophoretic heterogeneity probably stems from (i) the different pattern of cysteine oxidations that can modify the protein mobility; (ii) the different amount and quality of cysteine oxidations in various molecules (“redox isomers”).
The gel-digestion procedure shows some disadvantages: (i) larger peptides can get trapped between the gel meshes and lost during the extraction phase of the peptides from the gel; (ii) the electrophoretic procedure itself could damage the samples and alter the redox state of the sulfur amino acids (due to possible over heating generated by the applied voltage and to the presence of residual quantities of the catalysts used for the polyacrylamide polymerization). Furthermore, electrophoresis requires a relatively high amount of sample and the utilization of dyes and detergents. These last molecules could interfere with subsequent MS analyses because these compounds are difficult to eliminate from the sample.
2-DE could potentially represent a useful alternative to 1-DE to improve the separation of VDAC isoforms, but its utilization presents other problems. Actually, this kind of proteins has been under-represented in 2-DE gels due to difficulties in extracting and solubilizing them in the isoelectric focusing sample buffer. In fact, the most effective solubilizing agent for highly hydrophobic membrane proteins is SDS, but this detergent is incompatible with 2-DE. In addition to the difficulties in entering IPG (immobilized pH gradient) gels, membrane proteins tend to precipitate at their isoelectric point during IEF. Furthermore, their tendency to absorb the IPG matrix prevents their migration into the SDS-PAGE gel.
An improvement in the rVDAC3 mass spectrometric analysis was obtained following the introduction of a gel-free shotgun proteomic approach [41]. According to this procedure, to avoid any possible artefact due to air exposure and manipulations, reduction/alkylation was carried out before VDACs purification from the mitochondria. Afterwards, all the proteins present in the HTP eluate, without previous electrophoretic separation, were purified from non-protein contaminating molecules with the PlusOne 2-D Clean-Up kit, and the desalted protein pellet was then re-dissolved in ammonium bicarbonate containing RapiGest SF to improve the solubility. In fact, this surfactant makes the proteins more susceptible to enzymatic cleavage without modifying the sample or inhibiting endoprotease activity. Furthermore, the RapiGest SF is compatible with enzymes such as trypsin or chymotrypsin and does not influence subsequent MS analysis because it can be easily removed in acidic conditions.
Separate aliquots of reduced and alkylated proteins were then subjected to digestion with modified porcine trypsin and chymotrypsin. In this experiment, every protein in the HTP eluate was digested, producing a very complex peptide mixture, which was finally analyzed by nUHPLC/HR nESI-MS/MS.
The new “in solution-digestion” protocol associated with nUHPLC/HR ESI-MS/MS allowed to extend the coverage of the rat and human VDACs sequences with respect to that obtained with the previous procedure [43], so that it was possible to completely cover the rat and human VDAC1 sequences and almost completely the rat and human VDAC2 and VDAC3 sequences [22][41][44]. It should be noted that the short regions not identified in VDAC2 and VDAC3 correspond to small tryptic or chymotryptic peptides or even to single amino acids, which cannot be detected in LC/MS analysis.
Moreover, by means of this new procedure a detailed characterization of PTMs of the three VDACs was obtained (see next paragraphs).
2.2. Mass Spectrometry Analysis of Post-Translational Modifications
The mammalian proteome is vastly more complex than the related genome. The reasons for this difference reside both in the molecular mechanisms that allow a single gene to encode for multiple proteins (genomic recombination, transcription initiation at alternative promoters, differential transcription termination, and alternative splicing of the transcript) and in the post-translational modifications (PTMs) which represent a wide range of chemical changes that proteins can undergo after synthesis. They include the specific cleavage of protein precursors, the covalent addition or removal of low-molecular weight groups (i.e., acetylation, glycosylation, hydroxylation, phosphorylation, ubiquitination) and the formation of disulfide bonds or other redox modifications [45][46][47].
PTMs play crucial roles in cell biology since they can change protein physical or chemical properties, activity, localization, and/or stability. Traditionally, PTMs have been identified by Edman degradation, amino acid analysis, isotopic labeling, or immunochemistry. Within recent years, MS has proven to be extremely useful in PTM discovery. Post-translationally modified amino acids always have a different molecular mass than the original, unmodified residues and this mass increment or deficit is usually the basis for the detection and characterization of PTM by MS (commonly by LC-ESI-MS/MS).
MS has several advantages for characterization of PTMs, including (i) very high sensitivity; (ii) discovery of novel PTMs; (iii) ability to identify PTMs and the modified sites, even in complex protein mixtures; and (iv) ability to quantify the relative changes in PTM occupancy at distinct sites. None of the other techniques provide all these features, so the greater majority of the known PTMs have been described by MS [48].
To improve sensibility, several methods have been developed to enrich the samples in proteins or peptides with specific PTMs prior to MS/MS analysis, such as anti-pY antibodies, IMAC (immobilized metal affinity chromatography) and TiO2 for phosphorylation [49][50], affinity capture with lectins for glycosylated proteins [51], and resin coupled with anti-acetyl-lysine for acetylated proteins [52]. Although, as previously described, isolation of single isoforms of VDACs cannot be obtained, application of combined HPLC and high-resolution ESI-MSMS analysis has resulted in the identification of several PTM in these proteins. In the following, a summary of the MS-based PTMs characterized in VDACs is reported and the respective biological significance discussed. These results are resumed in Table 1 and Figure 1.
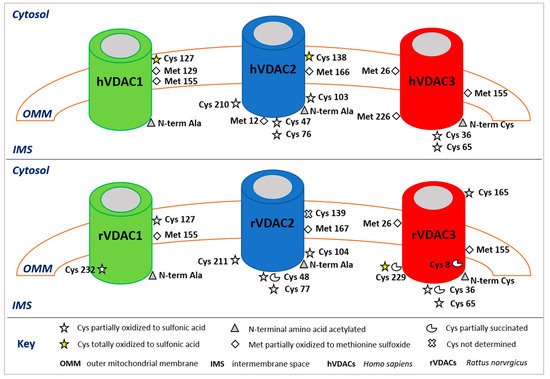
Figure 1. Post-translational modifications of human (upper panel) and rattus (lower panel) VDAC isoforms. The image shows only the modified amino acids and their positions with respect to the cytosol, the outer mitochondrial membrane (OMM), and the intermembrane space (IMS). In the rVDAC1 Cys232 faces the aqueous inside of the pore; in the rVDAC3 Cys8 is located inside of the pore.
Table 1. Post-translational modifications in VDAC isoforms obtained using mass spectrometry. PTM type, mass shift (Da), source of the sample, modified residue, MS method and relative reference are reported. Studies are described by listing first author + year.
| ISOFORM | PTM Type | ΔMass (Da) | Source | Residue | Method | Study |
|---|---|---|---|---|---|---|
| VDAC1 | Protein N-terminal acetylation | 42.0106 | Rat liver | Ala 2 | nUHPLC/high resolution nESI-MS/MS in a Q-QT-qIT MS |
Saletti et al., 2018 |
| HAP1 cells | Ala 2 | Pittalà et al., 2020 | ||||
| Acetylation | 42.0106 | Mouse liver | Lys 33, 41, 74, 234 | nHPLC MS/MS in an LTQ MS | Kim et al., 2006 | |
| Lys 41, 122, 132 | nHPLC MS/MS in an LTQ 2D ion-trap MS |
Schwer et al., 2009 | ||||
| Mouse liver and heart | Lys 237 | UPLC Velos-FT MS | Yang et al., 2011 | |||
| Human liver | Lys 28 | LC/LC-MS/MS in an FTICR/MS | Zhao et al., 2010 | |||
| Oxidation | 15.9949 | Rat liver | Met 155 | LC/LC-MS/MS in an FTICR/MS | Guan et al., 2003 | |
| nUHPLC/high resolution nESI-MS/MS in a Q-QT-qIT MS |
Saletti et al., 2018 | |||||
| HAP1 cells | Met 129, 155 | Pittalà et al., 2020 | ||||
| Trioxidation | 47.9847 | Rat liver | Cys 127, 232 | Saletti et al., 2018 | ||
| HAP1 cells | Cys 127 | Pittalà et al., 2020 | ||||
| Phosphorylation | 79.9663 | Rat liver | Ser 12, 136 | HPLC MS/MS in an LTQ MS | Distler et al., 2007 | |
| Mouse liver | Ser 117 | nHPLC MS/MS in an LTQ MS | Lee et al., 2007 | |||
| HeLa cells | Ser 101, 102, 104, Thr 107 |
nHPLC MS/MS in an LTQ-Orbitrap MS |
Olsen et al., 2006 | |||
| Mouse brain | Tyr 80, 208 | LC-MS/MS in an LTQ FT MS | Ballif et al., 2008 | |||
| VDAC2 | Protein N-terminal acetylation | 42.0106 | Rat liver | Ala 2 | nUHPLC/high resolution nESI-MS/MS in a Q-QT-qIT MS |
Saletti et al., 2018 |
| HAP1 cells | Ala 2 | Pittalà et al., 2020 | ||||
| Acetylation | 42.0106 | Mouse liver | Lys 32, 75 | nHPLC MS/MS in an LTQ MS | Kim et al., 2006 | |
| Lys 121 | nHPLC MS/MS in an LTQ 2D ion-trap MS |
Schwer et al., 2009 | ||||
| Oxidation | 15.9949 | Rat liver | Met 167 | nUHPLC/high resolution nESI-MS/MS in a Q-QT-qIT MS |
Saletti et al., 2018 | |
| HAP1 cells | Met 12, 166 | Pittalà et al., 2020 | ||||
| Trioxidation | 47.9847 | Rat liver | Cys 48, 77, 104, 211 | Saletti et al., 2018 | ||
| HAP1 cells | Cys 47, 76, 103, 138, 210 | Pittalà et al., 2020 | ||||
| Succination | 116.0110 | Mouse brain | Cys 48, 77 | LC-nESI-MS/MS in an LTQ-Orbitrap MS |
Piroli et al., 2016 | |
| Rat liver | Cys 48 | nUHPLC/high resolution nESI-MS/MS in a Q-QT-qIT MS |
Saletti et al., 2018 | |||
| Phosphorylation | 79.9663 | HeLa cells | Ser 115, Thr 118 | nHPLC MS/MS in an LTQ-Orbitrap MS |
Olsen et al., 2006 | |
| Rat liver | Thr 109 | SCX-RP-MS/MS in an LTQ-Orbitrap MS |
Deng et al., 2010 | |||
| Rat liver | Tyr 237 | HPLC MS/MS in an LTQ MS | Distler et al., 2007 | |||
| Mouse brain | Tyr 207 | LC-MS/MS in an LTQ FT MS | Ballif et al., 2008 | |||
| VDAC3 | Protein N-terminal acetylation | 42.0106 | Rat liver | Cys 2 | nUHPLC/high resolution nESI-MS/MS in a Q-QT-qIT MS |
Saletti et al., 2016 |
| HAP1 cells | Cys 2 | Pittalà et al., 2020 | ||||
| Acetylation | 42.0106 | Mouse liver | Lys 20, 61, 226 | nHPLC MS/MS in an LTQ MS | Kim et al., 2006 | |
| Lys 63, 109 | nHPLC MS/MS in an LTQ 2D ion-trap MS |
Schwer et al., 2009 | ||||
| Human liver | Lys 28 | LC/LC-MS/MS in an FTICR-MS | Zhao et al., 2010 | |||
| Oxidation | 15.9949 | Rat liver | Met 26, 155 | nUHPLC/high resolution nESI-MS/MS in a Q-QT-qIT MS |
Saletti et al., 2016 | |
| HAP1 cells | Met 26, 155, 226 | Pittalà et al., 2020 | ||||
| Trioxidation | 47.9847 | Rat liver | Cys 36, 65, 165, 229 | Saletti et al., 2016 | ||
| HAP1 cells | Cys 36, 65 | Pittalà et al., 2020 | ||||
| Succination | 116.0110 | Rat liver | Cys 8, 36, 229 | Saletti et al., 2018 | ||
| Phosphorylation | 79.9663 | Rat liver | Ser 241, Thr 33 | HPLC MS/MS in an LTQ MS | Distler et al., 2007 | |
| Mouse brain | Tyr 49 | LC-MS/MS in an LTQ FT MS | Ballif et al., 2008 |
References
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochon-drial protein regulating cell life and death. Mol. Aspects Med. 2010, 31, 227–285.
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476.
- Young, M.J.; Bay, D.C.; Hausner, G.; Court, D.A. The evolutionary history of mitochondrial porins. BMC Evol. Biol. 2007, 7, 31.
- De Pinto, V.; Reina, S.; Gupta, A.; Messina, A.; Mahalakshmi, R. Role of cysteines in mammalian VDAC isoforms’ function. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1219–1227.
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 2008, 321, 1206–1210.
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375.
- Ujwal, R.; Cascio, D.; Colletier, J.P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse VDAC1 at 2.3 angstrom resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747.
- Schredelseker, J.; Paz, A.; Lopez, C.J.; Altenbach, C.; Leung, C.S.; Drexler, M.K.; Chen, J.N.; Hubbell, W.L.; Abramson, J. High resolution structure and double electron-electron resonance of the zebrafish voltage-dependent anion channel 2 reveal an oligomeric population. J. Biol. Chem. 2014, 289, 12566–12577.
- Amodeo, G.F.; Scorciapino, M.A.; Messina, A.; De Pinto, V.; Ceccarelli, M. Charged residues distribution modulates selec-tivity on the open state of human isoforms of the voltage dependent anion-selective channel. PLoS ONE 2014, 9, e103879.
- De Pinto, V.; Guarino, F.; Guarnera, A.; Messina, A.; Reina, S.; Tomasello, M.F.; Palermo, V.; Mazzoni, C. Characterization of human VDAC isoforms: A peculiar function for VDAC3? Biochim. Biophys. Acta-Bioenergetics 2010, 1797, 1268-1275.
- Naghdi, S.; Várnai, P.; Hajnóczky, G. Motifs of VDAC2 required for mitochondrial Bak import and tBid-induced apoptosis. Proc. Natl. Acad. Sci. USA 2015, 112, E5590–E5599.
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell stress 2017, 1, 1.
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348.
- Kerner, J.; Lee, K.; Tandler, B.; Hoppel, C.L. VDAC proteomics: Post-translation modifications. Biochim. Biophys. Acta 2012, 1818, 1520–1525.
- Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of vdac1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485.
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The vdac1 n-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122, 1906–1916.
- Shi, Y.; Chen, J.;Weng, C.; Chen, R.; Zheng, Y.; Chen, Q.; Tang, H. Identification of the protein-protein contact site and inter-action mode of human vdac1 with bcl-2 family proteins. Biochem. Biophys. Res. Commun. 2003, 305, 989–996.
- Naghdi, S.; Hajnóczky, G. VDAC2-specific cellular functions and the underlying structure. Biochim. Biophys. Acta 2016, 1863, 2503–2514.
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochon-drion 2012, 12, 24–34.
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490.
- De Pinto, V.; al Jamal, J.A.; Palmieri, F. Location of the dicyclohexylcarbodiimide-reactive glutamate residue in the bovine heart mitochondrial porin. J. Biol. Chem. 1993, 268, 12977–12982.
- Pittalà, M.G.G.; Saletti, R.; Reina, S.; Cunsolo, V.; De Pinto, V.; Foti, S. A High Resolution Mass Spectrometry Study Reveals the Potential of Disulfide Formation in Human Mitochondrial Voltage-Dependent Anion Selective Channel Isoforms (hVDACs). Int. J. Mol. Sci. 2020, 21, 1468.
- Budelier, M.M.; Cheng, W.W.L.; Bergdoll, L.; Chen, Z.W.; Janetka, J.W.; Abramson, J.; Krishnan, K.; Mydock-McGrane, L.; Covey, D.F.; Whitelegge, J.P.; et al. Photoaffinity labeling with cholesterol analogues precisely maps a cholesterol-binding site in voltage-dependent anion channel-1. J. Biol. Chem. 2017, 292, 9294–9304.
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. Tspo interacts with vdac1 and triggers a ros-mediated inhibition of mitochondrial quality control. Autophagy 2015, 10, 2279–2296.
- Mueller, B.K.; Subramaniam, S.; Senes, A. A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Cα-H hydrogen bonds. Proc. Natl. Acad. Sci. USA 2014, 111, E888–E895.
- Bernardi, P.; Di Lisa, F.; Fogolari, F.; Lippe, G. From ATP to PTP and back: A dual function for the mitochondrial ATP syn-thase. Circ. Res. 2015, 116, 1850–1862.
- Reina, S.; Guarino, F.; Magrì, A.; De Pinto, V. VDAC3 as a potential marker of mitochondrial status is involved in cancer and pathology. Front. Oncol. 2016, 6, 264.
- Magrì, A.; Messina, A. Interactions of VDAC with Proteins Involved in Neurodegenerative Aggregation: An Opportunity for Advancement on Therapeutic Molecules. Curr. Med. Chem. 2017, 24, 4470–4487.
- Sun, Y.; Vashisht, A.A.; Tchieu, J.; Wohlschlegel, J.A.; Dreier, L. Voltage-dependent Anion Channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 2012, 287, 40652–40660.
- Sheikh, S.; Safia; Haque, E.; Snober, S.M. Neurodegenerative diseases: Multifactorial conformational diseases and their therapeutic interventions. J. Neurodegen. Dis. 2013, 2013, 563481.
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The voltage-dependent anion channel 1 mediates amyloid beta toxicity and represents a potential target for Alzheimer's disease therapy. J. Biol. Chem. 2015, 290, 30670–30683.
- Hemachandra Reddy, P. Is the mitochondrial outer membrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta 2013, 1832, 67–75.
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein shows high affinity interaction with Voltage-dependent Anion Channel, suggesting mechanisms of mitochon-drial regulation and toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477.
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587.
- Magrì, A.; Belfiore, R.; Reina, S.; Tomasello, M.F.; Di Rosa, M.C.; Guarino, F.; Leggio, L.; De Pinto, V.; Messina, A. Hexoki-nase I N-terminal based peptide prevents the VDAC1-SOD1G93A interaction and re-establishes ALS cell viability. Sci. Rep. 2016, 6, 34802.
- Harada, T.; Sada, R.; Osugi, Y.; Matsumoto, S.; Matsuda, T.; Hayashi-Nishino, M.; Nagai, T.; Harada, A.; Kikuchi, A. Pal-mitoylated CKAP4 regulates mitochondrial functions through an interaction with VDAC2 at ER–mitochondria contact sites. J. Cell Sci. 2020, 133, jcs249045.
- Zhong, Y.; Tang, X.; Sheng, X.; Xing, J.; Zhan, W. Voltage-Dependent Anion Channel Protein 2 (VDAC2) and Receptor of Ac-tivated Protein C Kinase 1 (RACK1) Act as Functional Receptors for Lymphocystis Disease Virus Infection. J. Virol. 2019, 93, e00122-19.
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; O’Connell, D.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; Chen, W. Nedd4 ubiq-uitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 433.
- Messina, A.; Reina, S.; Guarino, F.; Magrì, A.; Tomasello, F.; Clark, R.E.; Ramsayc, R.R.; De Pinto, V. Live cell interactome of the human voltage dependent anion channel 3 (VDAC3) revealed in HeLa cells by affinity purification tag technique. Mol. BioSyst. 2014, 10, 2134–2145.
- Abrecht, H.; Wattiez, R.; Ruysschaert, J.M.; Homblé, F. Purification and characterization of two Voltage-Dependent Anion Channel Isoforms from plant seeds. Plant Physiol. 2000, 124, 1181–1190.
- Saletti, R.; Reina, S.; Pittalà, M.G.G.; Belfiore, R.; Cunsolo, V.; Messina, A.; De Pinto, V.; Foti, S. High resolution mass spec-trometry characterization of the oxidation pattern of methionine and cysteine residues in rat liver mitochondria Volt-age-Dependent Anion selective Channel 3 (VDAC3). Biochim. Biophys. Acta-Biomembr. 2017, 1859, 301–311.
- De Pinto, V.; Prezioso, G.; Palmieri, F. A simple and rapid method for the purification of the mitochondrial porin from mammalian tissues. Biochim. Biophys. Acta 1987, 905, 499–502.
- Distler, A.M.; Kerner, J.; Peterman, S.M.; Hoppel, C.L. A targeted proteomic approach for the analysis of rat liver mito-chondrial outer membrane proteins with extensive sequence coverage. Anal. Biochem. 2006, 356, 18–29.
- Saletti, R.; Reina, S.; Pittalà, M.G.G.; Magrì, A.; Cunsolo, V.; Foti, S.; De Pinto, V. Post-translational modifications of VDAC1 and VDAC2 cysteines from rat liver mitochondria. Biochim. Biophys. Acta-Bioenerg. 2018, 1859, 806–816.
- Wang, Y.; Peterson, S.; Loring, J. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 24, 143–160.
- Duan, G.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 2015, 11, e1004049.
- Reina, S.; Pittalà, M.G.G.; Guarino, F.; Messina, A.; De Pinto, V.; Foti, S.; Saletti, R. Cysteine oxidations in mitochondrial membrane proteins: The case of VDAC isoforms in mammals. Front. Cell Dev. Biol. 2020, 8, 397.
- Jensen, O.N. Modification-specific proteomics: Characterization of posttranslational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 2004, 8, 33–41.
- Corthals, G.L.; Aebersold, R.; Goodlett, D.R. Identification of phosphorylation sites using microimmobilized metal affinity chromatography. Meth. Enzymol. 2005, 405, 66–81.
- Larsen, M.R.; Thingholm, T.E.; Jensen, O.N.; Roepstorff, P.; Jørgensen, T.J. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 2005, 4, 873–886.
- Yang, Z.; Hancock, W.S. Approach to the comprehensive analysis of glycoproteins isolated from human serum using a mul-ti-lectin affinity column. J. Chromatogr. A 2004, 1053, 79–88.
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell. 2006, 23, 607–618.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
4 times
(View History)
Update Date:
19 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No