Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Konstantin Lyamzaev | + 3449 word(s) | 3449 | 2021-12-20 04:15:52 | | | |
| 2 | Bruce Ren | Meta information modification | 3449 | 2021-12-21 01:47:18 | | | | |
| 3 | Bruce Ren | Meta information modification | 3449 | 2021-12-22 08:54:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lyamzaev, K. Innate Immunity Promotes Programmed Death of Individual Organisms. Encyclopedia. Available online: https://encyclopedia.pub/entry/17343 (accessed on 24 July 2026).
Lyamzaev K. Innate Immunity Promotes Programmed Death of Individual Organisms. Encyclopedia. Available at: https://encyclopedia.pub/entry/17343. Accessed July 24, 2026.
Lyamzaev, Konstantin. "Innate Immunity Promotes Programmed Death of Individual Organisms" Encyclopedia, https://encyclopedia.pub/entry/17343 (accessed July 24, 2026).
Lyamzaev, K. (2021, December 20). Innate Immunity Promotes Programmed Death of Individual Organisms. In Encyclopedia. https://encyclopedia.pub/entry/17343
Lyamzaev, Konstantin. "Innate Immunity Promotes Programmed Death of Individual Organisms." Encyclopedia. Web. 20 December, 2021.
Copy Citation
Over-activation of innate immunity in response to bacterial or viral (including COVID-19) infections, massive trauma, or ischemia/reperfusion often causes severe illness and death. We suggest that such severe outcomes may be manifestations of an altruistic suicidal strategy protecting the entire population from the spread of pathogens and from dangerous pathologies rather than aberrant hyperstimulation of defense responses.
pathogen-associated molecular patterns (PAMPs)
damage-associated molecular patterns (DAMPs)
mitochondrially-targeted antioxidants
inflammation
inflammasome
programmed death
phenoptosis
COVID-19
1. Introduction
The concept of altruistic programmed death of whole organisms was proposed by Vladimir Skulachev more than two decades ago [1][2]. It was based on a suggestion that the death of individuals, if programmed, may be a subject to Darwinian selection and contribute to inclusive fitness. This suggestion expanded the original idea of inclusive fitness, also known as kin selection, which initially was about promoting reproduction and survival of genetic relatives only via social behavior [3]. In addition to this concept, Skulachev suggested that Darwinian selection may have shaped the mechanisms “of clearing a kin community of organisms or a population from individuals who have become unwholesome for this community”. He suggested to call this type of kin selection “phenoptosis”. Skulachev suggested that “the septic shock and stress-induced ischemic diseases of brain and heart” could be considered as the examples of phenoptosis [1]. At the same time, “slow phenoptosis” was proposed as an equivalent term for programmed aging [1][2][4].
The concept of phenoptosis has received support in experimental [5] and computer studies of Caenorhabditis elegans [6][7]. A cold shock followed by rewarming induced expression of genes of proteases, which promoted the programmed organismic death [5]. In the model of a clonal population of C. elegans that subsists on spatially limited food sources, simulations have shown that shorter lifespan can increase the colony fitness [6][7]. Presumably, phenoptosis, by reducing the futile food consumption by elderly or stressed worms leads to adaptive benefits for the colony. This concept is also fully consistent with the recently published results of mathematical modeling [8]. The authors demonstrated that in a population of short-lived individuals, the spread of infection is limited, and the pathogen clearance is more efficient than in population in which members live longer.
For diverse types of programmed cell death (PCD), such as apoptosis, necroptosis, and pyroptosis, specific execution mechanisms were identified [9]. Therefore, to categorize phenoptosis as a specific bona fide program, a specific biochemical execution mechanism(s) has to be identified as well.
2. Innate Immunity, Pathogen-Associated Molecular Patterns, and Pattern Recognition Receptors
The basic principle of innate immunity, as formulated by C. A. Janeway Jr [10], states that diverse invading pathogens are immediately recognized by molecules that are broadly shared by pathogens, such as glycans, N-formylated peptides, flagellin, and nucleic acids, but are distinguishable from host molecules. These molecules are collectively referred to as pathogen-associated molecular patterns (PAMPs). Some PAMPs are shared by viruses and diverse groups of microorganisms, including non-pathogenic ones.
PAMPs are recognized by pattern recognition receptors (PRRs). The first of the discovered PRRs are Toll-like receptors (TLR), of which there are at least 10 in humans [11][12]. Later several further classes of PRRs were discovered including C-type lectin receptors (CLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors, NOD (nucleotide-binding oligomerization domain)-like receptors (NLRs), and cytosolic DNA sensors [13][14].
TLRs are located in the plasma membrane or in the membranes of intracellular compartments (endoplasmic reticulum, endosomes, lysosomes), recognizing a wide variety of substances of microbial and viral origin [11][15].
CLRs make a large group of transmembrane or soluble receptors expressed primarily by myeloid cells. They recognize carbohydrates (glycans) on the surface of bacteria, fungi, helminths, and viruses [16].
The RIG-I-like receptors are intracellular viral RNA receptors. This family includes RIG-I that recognizes RIG-I circular RNA and short (≤300 bp) dsRNAs, MDA5 that senses long dsRNAs, and LGP2 that binds dsRNA and regulates signaling by RIG-I and MDA5 [17].
The NLR family has 22 members in humans, which are cytosolic sensors expressed in various immune and non-immune cells [18]. The NOD1 and NOD2 receptors recognize the degradation products of bacterial peptidoglycan, in particular muramyl dipeptide, which is characteristic of numerous pathogenic bacteria. The NLRP1, NLRP3, and NLRC4 receptors, in response to pro-inflammatory stimuli, form inflammasomes, large multiprotein complexes in which caspase-1 is activated to convert the precursors of interleukin 1β (pro-IL-1β) and interleukin 18 (pro-IL-18) into their active forms and to stimulate their release [18][19][20]. In addition to NLRs, the inflammasome formation can be induced by the interferon-inducible protein AIM2, the cytosolic DNA sensor [21].
The main PRR that recognizes the cytosolic DNA is the cyclic guanosine monophosphate adenosine monophosphate synthase (cGAS) that, by using STING (cGAS stimulator of interferon genes), stimulates the production of type-I interferon (IFN-I) [22]. Several other soluble receptors for cytosolic DNA and RNA have recently been identified [21]. All these sensors recognize DNA that can emerge during the life cycle of intracellular pathogens including viruses, bacteria, and parasites.
3. Damage Associated Molecular Patterns (DAMPs) and Their Receptors
In addition to PAMPs, the same TLRs, as well as some other PRRs, recognize damage-associated molecular patterns (DAMPs), which are endogenous molecules released from damaged cells, see [23][24] and Figure 1. Various types of DAMPs and their role in inflammation were extensively reviewed [25][26].
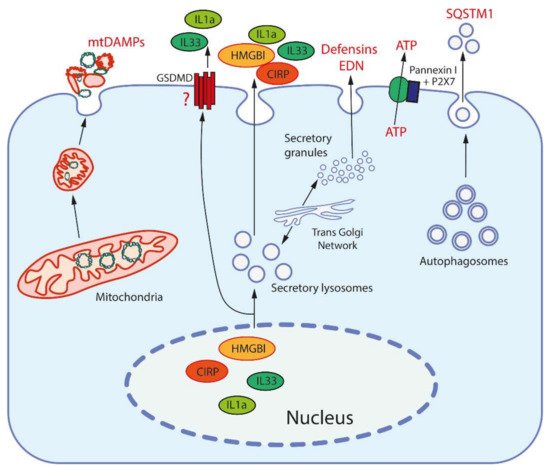
Figure 1. Release mechanisms for some DAMPs. High-mobility group protein B1 (HMGB1) and cold-inducible RNA-binding protein (CIRP) are released from the nucleus after modification to be entrapped and secreted by secretory lysosomes (which are produced by the trans-Golgi network). Interleukins 1a and 33 (IL1a, IL33) are also secreted by secretory lysosomes, but may be also released through the pores formed by gasdermin D (GSDMD). Mitochondria, after fragmentation, can be extruded from cells by an unknown mechanism(s). Secretory granules, as produced by the trans-Golgi network, release defensins and eosinophil-derived neurotoxin (EDN) by exocytosis. ATP is released through the pore formed by pannexin I complexed with the purinergic receptor P2X7. Sequestosome-1 (SQSTM1/h62) is an autophagosome receptor that can be released during secretory autophagy.
The concept of DAMPs stems from the “danger model” of immunity by P. Matzinger [23]. In contrast to the traditional “self versus non-self” recognition principle, she proposed that the immune system recognizes danger signals regardless of their origin. Initially, this hypothesis concerned adaptive immunity [23], but later the same principle was applied to innate immunity [24].
DAMPs can directly initiate at least two forms of PCD (pyroptosis and NETosis), which are involved in the pathogenesis of sepsis, as well as many other diseases associated with aseptic inflammation. Pyroptosis is a form of cell death that is accompanied by activation of a pore-forming protein gasdermin D by inflammatory caspases [27]. NETosis (where NET is deciphered as neutrophil extracellular traps) is a specific form of cell death that is characterized by the release of weblike DNA structures decorated with histones and antimicrobial proteins by neutrophils [28]. Cytokines produced in response to DAMP can also initiate other forms of PCD, such as apoptosis and necroptosis, which also contribute to various pathologies.
Today, a large number of DAMPs of various chemical nature and origins have been identified (see Table 1), and this list is expanding rapidly. The most important and well characterized DAMPs, namely the high-mobility group protein B1 (HMGB1) and cold-inducible RNA-binding protein (CIRP), function as chaperons in the nucleus [29][30]. Two interleukins of the IL-1 family, IL-1α and IL-33, are nuclear proteins capable of regulating expression. Outside the cell, they are recognized not by PRRs, but by the receptors of the IL1R family (IL-R1 and ST2, respectively), which transmit signals using the same MyD88 adapter as some TLRs [31]. Some other cytokines, such as thymic stromal lymphopoietin (TSLP) and IL-25, are also considered DAMPs although their intracellular functions have not been defined [31]. Expression of all these DAMPs is stimulated by various stresses and by mediators of inflammation.
Table 1. Typical damage-associated molecular patterns (DAMPs).
| # | Origin | DAMPs | Receptors | Ref |
|---|---|---|---|---|
| 1 | Nucleus | HMGB1 | TLR2, TLR4, RAGE | [29] |
| CIRP | TLR4, TREM-1 | [30] | ||
| Histones | TLR2, TLR4 | [32] | ||
| SAP130 | Mincle | [26] | ||
| IL-1α | IL1R1 | [31] | ||
| IL-33 | ST2 | [31] | ||
| DNA | cGAS, AIM2, RAGE, IFI16 | [21] | ||
| 2 | Cytosol | S100 proteins | TLR2, TLR4, RAGE | [31] |
| HSPs | TLR2, TLR4, CD91 | [33] | ||
| F-Actin | DNGR-1, TREM1 | [34] | ||
| Cyclophilin A | CD147 | [35] | ||
| Peroxiredoxin 1 | TLR4 | [36] | ||
| Oxidized hemoglobin, heme | TLR4 | [37] | ||
| Amyloid β | TLR4 | [38] | ||
| ATP, ADP | P2X7R, P2Y2R, P2Y12R, | [39] | ||
| Uric acid | TREM-1, TLR2, TLR4, P2X7, NLRP3 | [26] | ||
| mRNA | TLR3 | [26] | ||
| microRNAs | TLR7 | [26] | ||
| SNAPIN | TLR2 | [26] | ||
| AGEs | RAGE | [26] | ||
| 3 | Mitochondria | Formyl peptides | FPR1 | [40] |
| mtDNA | TLR9, NLRP3 | [40] | ||
| Cardiolipin | NLRP3, TREM2 | [41] | ||
| Cytochrome c | TLR4 | [42] | ||
| Oxygenated mitochondrial fatty acids | TRL4 | [43][44] | ||
| TFAM | RAGE | [40] | ||
| 4 | ER, secretory granules, autophagosomes | Defensins | TLR4 | [45] |
| Cathelicidins | P2X7, FPR2 | [45] | ||
| Eosinophil-derived neurotoxin | TLR2 | [46] | ||
| Granulisin | TLR4 | [47] | ||
| Calreticulin | CD91 | [48] | ||
| Gp96 | TLR2, TLR4, CD91 | [48] | ||
| Sequestosome-1 (SQSTM1 or p62) | INSR | [49] | ||
| 5 | Extracellular matrix | Heparan sulphate, versican, aggrecan | TLR4 | [50] |
| Proteoglycans (biglycan, decorin, etc.) | TLR2, TLR4, CD14, NLRP3 | [50] | ||
| Tenascin-C | TLR4 | [50] | ||
| Fibrinogen | TLR4 | [50] | ||
| Fibronectin | TLR2, TLR4 | [50] | ||
| Low molecular weight hyaluronan | TLR2, TLR4, NLRP3 | [50] | ||
| 6 | Tumor cells | Annexin A1 | FPR1 | [51] |
| PAUF | TLR4 | [51] | ||
| API5 | TLR4 | [51] | ||
| Rps-3 | TLR4 | [51] |
Cytosolic DAMPs, such as S100 proteins that regulate Ca2+ signaling [31], actin, a component of cytoskeleton [34], peptidyl prolyl isomerase cyclophilin A [35], antioxidant protein peroxiredoxin 1 [36], and heat shock proteins (HSPs) [33] are among the most abundant proteins. Oxidized hemoglobin and heme, as released from red blood cells upon hemolysis, also act as DAMPs [37].
Secretory granules are another important source of DAMPs; these are antimicrobial peptides (defensins and LL37) [45], eosinophil-derived neurotoxin (EDN) [46], and granulisin, which accumulates in the granules of human cytotoxic T lymphocytes and natural killer cells [47].
Molecules of ATP [39] and uric acid (UA) [52] are the most important low molecular weight metabolites involved in the activation of inflammation upon release from the cell. In addition, various components of the extracellular matrix (ECM), including proteoglycans, soluble glycoproteins, low molecular weight hyaluronan, and heparan sulfate, are capable of acting as DAMPs [50][40].
Mitochondria are regarded as an important source of DAMPs (mtDAMPs) primarily due to their bacterial origin. Mitochondria-synthesized proteins are N-formylated, and mitochondrial DNA (mtDNA) contains insufficiently methylated CpG regions, similarly to the corresponding bacterial components which are recognized as PAMPs. One example of mtDAMPs in common with some bacteria is cytochrome c [42]. In contrast, the transcription factor A (TFAM) is an example of a mtDAMPs that appears not to have a bacterial counterpart [40]. Almost the entire list of known DAMPs can be released from tumors and contribute to immunogenic cell death (ICD) associated with an adaptive immune response [51]. Some of the DAMPs (not all), including those that are predominantly released from tumors, are listed in Table 1.
DAMPs can be released from cells via active and passive pathways (Figure 1). Passive release requires the rupture of the plasma membrane and occurs during pyroptosis, necroptosis, or necrosis. Both nuclear DNA and histones can be released from leucocytes in form of decondensed chromatin forming extracellular traps (ETs; NETs in the case of neutrophils). Interestingly, some DAMPs (HMGB1, CIRP) can induce release of NETs, which leads to the formation of an amplification loop [53].
Active release of nuclear DAMPs, such as HMGB1 and CIRP, requires their posttranslational modification in order to leave the nucleus, and subsequently occurs through exocytosis of secretory lysosomes [29][30]. The same exocytotic mechanism participates in active release of cytosolic and mitochondrial DAMPs. ATP can be released not only from damaged cells, but also through the channel formed by the P2X purinoreceptor 7 (P2X7 receptor) together with pannexins [39]. Uric acid excretion is largely regulated by urate transporters in the renal tubules [52]. In general, it can be assumed that the release of the most important DAMPs may be mediated by specific mechanisms in living cells (Figure 1).
Initially, it was thought that DAMPs are only recognized by pattern recognition receptors (PPRs), but later the receptors for advanced glycation end products (RAGE), triggering receptor expressed on myeloid cells 1 (TREM-1), CD91 and purinergic receptors were also shown to participate in the DAMP recognition [25][26]. It has recently been reported that sequestosome 1 (SQSTM1/p62), a known selective autophagy receptor released by macrophages and monocytes, is recognized by the insulin receptor (INSR) to stimulate inflammation [49]. In addition, formyl peptide receptor 1 (FPR1), is a G protein-coupled receptor that recognizes N-formylmethionine-containing peptides [54].
Some DAMPs can be recognized without release from the cell. MtDNA, released from mitochondria into cytosol and oxidized, can stimulate the NLRP3 inflammasome activity [55]. Another intracellular DAMP is cardiolipin, a four-tail phospholipid which is located exclusively in the inner mitochondrial membrane. Under stress, cardiolipin is externalized on the mitochondrial surface and stimulates the assembly of the NLRP3 inflammasome [41].
Signal transduction from DAMP-sensing receptors uses some of signaling modules that are activated by elevated cytosolic levels of Ca2+ and reactive oxygen species (ROS). The main outcomes of DAMP-dependent signaling include NFκB and inflammasomes activation, followed by the production of inflammatory cytokines and chemokines, activation of mitogen-activated protein kinases (MAPK), and stimulation of interferon signaling (Figure 2), see [26] for a comprehensive review.
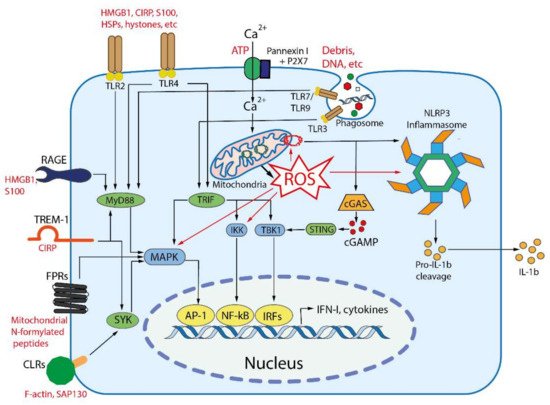
Figure 2. Main DAMP-sensing receptors and signaling. DAMPs can be recognized by membrane receptors, such as Toll- like receptors (TLRs), receptor for advanced glycation end products (RAGE), triggering receptor expressed on myeloid cells 1 (TREM-1), formyl peptide receptors (FPRs), C-type lectin receptors (CLRs), purinergic receptor P2X7, as well as by cytoplasmic receptors such as cyclic GMP-AMP synthase (cGAS) and NLR family pyrin domain containing 3 (NLRP3) inflammasome. Endogenous nuclear and mitochondrial DNA released into the cytoplasm activate cGAS, which produces cyclic GMP-AMP (cGAMP) that binds to stimulator of interferon genes (STING). Activated STING stimulates the kinases TBK1 and IKK, leading to the expression of type-I interferons (IFN-I) and inflammatory cytokines. TLR2 and TLR4 can be activated by a variety of extracellular DAMPs, whereas TLRs 3, 7, and 9 are activated by phagocytosed nucleic acids in endosomes. TLR-dependent signaling is mediated by the adaptor proteins: myeloid differentiation primary-response 88 (MyD88), and TIR-domain-containing adaptor inducing IFNβ (TRIF). The both adaptors activate mitogen-activated protein kinases (MAPKs) and IκB kinase (IKK), leading to activation of the transcription factors activator protein 1 (AP-1) and nuclear factor κB (NFκB), which stimulate the expression of inflammatory cytokines. TRIF also activates TANK binding kinase 1 (TBK1), which activates the interferon regulatory factors (IRFs) leading to the expression of type I interferon (IFN-I). RAGEs, which recognize HMGB1, S100 and some other DAMPs, also activate MyD88. TREM-1, which recognizes CIRP, activates spleen tyrosine kinase (SYK), that further stimulates MyD88, MAPKs and NFκB. Several C-type lectin receptors (CLRs) recognize F-actin and SAP130 and also activate SYK. Formyl peptide receptors (FPRs) are G-protein coupled receptors that recognize mitochondrial N-formylated peptides and stimulate several signaling pathways (including MAPKs) to activate neutrophils. P2X7 is ionotropic receptor that opens Ca2+ channel in response to extracellular ATP. An increase in the concentration of Ca2+ in cytoplasm modulates various signaling pathways and stimulates the production of reactive oxygen species (ROS) in mitochondria. Elevated ROS levels are critical for the activation of MAPKs, NFκB and the NLRP3 inflammasome. ROS-oxidized mitochondrial DNA is recognized by NLRP3.
In sum, DAMPs, as well as PAMPs, initiate an innate immune response, which involves the release of cytokines and other inflammatory mediators, activation of some specific defense mechanisms (for example, the production of extracellular traps), and induction of certain types of programmed cell death. Such a combination of organismic reactions is commonly called inflammation, which has been recently defined as “the innate immune response to harmful stimuli such as pathogens, injury, and tissue malfunction” [56]. This definition covers both acute (local or systemic) and chronic inflammatory processes.
4. Innate Immunity-Mediated Phenoptosis as a Common Cause of Human Mortality
In the previous sections we have emphasized that manifestations of innate immunity mechanisms in many pathological conditions led to death even when the pathological agents (viruses or bacteria) cannot cause death per se. We also showed that such fatalities could be prevented by interfering, in diverse ways, with the specific innate immunity regulatory pathways controlled by mitochondria. In addition, it was demonstrated that the probability of death outcome (from all causes) correlates with the presence of certain blood biomarkers most of which are related to inflammation.
Up to now, the above-described fatalities were attributed to the imperfection of the innate immunity system causing its overreaction. However, the respective mortality rates are very high, up to 50% in the case of sepsis, even in the best intensive care units around the world. High mortality is also inherent in various pathologies associated with local inflammation (ischemic and toxic lesions of the brain, heart, kidneys, liver, etc.), and in some pathologies where inflammation is not clearly expressed, but DAMP plays an important role in pathogenesis (including aforementioned neurodegenerative diseases). Such extremely high, seemingly wanton mortality must have an evolutionary cause. More likely is that (i) the high mortality, as caused by sepsis, sterile systemic inflammation, and some described severe infections, is a manifestation of phenoptosis that aims at cleansing the population of unwanted individuals, and (ii) innate immunity reactions are in a similar way involved in all the above-described cases because phenoptosis uses them as an executive mechanism. Involvement of the same innate immunity pathways, under certain circumstances leading to death, explains the similarity of symptoms in the generalized, pathogen-induced inflammation including sepsis and in sterile types of inflammation.
Cancers also fit into this framework. As early as in 1994, Sommer suggested that cancer “has a biological role in that it mediates evolutionary selection for a constant rate of germline mutation” (quoted from [57]) [1]. Cancer can be one of the most important mediators of negative selection, not only in the case of mutations (germline, as well as somatic), as discussed by Sommer [57], but also in pathologies related to systemic inflammation. Although it was repeatedly shown that inflammation can provoke cancer, inflammation plays a dual role in cancer [58]. On the one (dark) side, DAMPs released from tumors during progression or as a result of therapy can directly promote invasion and metastasis by interacting with PRRs expressed on tumor cells [59][60]. Moreover, DAMP-induced chronic inflammation in the tumor microenvironment attracts immunosuppressive cells such as M2 macrophages, myeloid suppressor cells and regulatory T cells, helping tumors escape immunosurveillance [61]. On the other (bright) side, DAMPs contribute to immunogenic tumor cell death caused by conventional therapy or modern cancer immunotherapy [51].
Generally, the existence of specific phenoptotic programs is indicated by the observations that such non-trivial symptoms as thrombosis and vascular leakage, hemorrhage, and organ failure were observed in relation to quite different causes, such as influenza, COVID-19, and some other respiratory viral infections, as well as in bacterial pneumonia, cholera and so on [62][63][64].
The existence of a distinct program gives a hope of preventing phenoptosis by interfering with the checkpoints of the program. Involvement of mitochondria as one of such checkpoints explains the aforementioned therapeutic effects of mitochondria-targeted antioxidants and very long chain 3-omega fatty acids in a variety of severe pathologies. It is noteworthy, that the impact of mitochondria-targeted antioxidants and very long chain 3-omega fatty acids is not just beneficial, but life-saving. Specifically, SkQ1-type antioxidants were the first to show a decrease in mortality in the kidney ischemia/reperfusion model [65], in models of pyelonephritis [66] and neonatal endotoxemia [67], as well as in a murine model of systemic inflammation induced by intravenous injection of TNF [68].
Identification of mitochondria as a phenoptosis checkpoint is also supported by a recent observation that high plasma level of very long omega-3 fatty acids lowered the risk for a fatal outcome in case of COVID-19 [69]. Most likely, omega-3 fatty acids prevented the formation of the NLRP3 inflammasome, which was activated in response to SARS-CoV2 and was found to be abundant in various tissues of postmortem patients upon autopsy [70].
In addition, the possible interruption of phenoptotic programs at the recognition stage of PAMPs or DAMPs could be a useful strategy to combat various life-threatening diseases. If so, the phenoptosis execution may be prevented by diminishing the level of certain DAMPs. And indeed, genetic knockouts, neutralizing antibodies, and pharmacological agents that inhibit DAMPs or prevent the activation of DAMP receptors such as TLRs, RAGE, NLRP3 inflammasome, and P2X7 ATP receptor, significantly attenuate the course and reduce mortality in animal models of systemic sterile inflammation [71]. The serum S100A8/A9 and HMGB1 levels were correlated an increased risk of lethal thrombosis [72]. Paquinimod, a specific inhibitor of S100A8/A9, reduced the number of aberrant neutrophils, eliminated lung damage, and protected mice in a lethal model of murine coronavirus infection [73]. Neutralizing antibodies against DAMPs significantly improved animal survival in models of endotoxemia and sepsis [53]. Small molecule inhibitors that prevent the secretion of HMGB1 and serum protein haptoglobin, which binds to extracellular HMGB1, have also been shown to have protective effects [74]. The important role of CIRP has been confirmed by the findings that CIRP−/− knockout mice are resistant to CLP sepsis [30]. In support of the role of extracellular histones in sepsis, several negatively charged molecules, including heparin, chondroitin sulfate, and high molecular weight hyaluronan, which can bind to histones of NETs were shown to protect against sepsis [75]. P2X receptor blockade or treatment with apyrase, that removes extracellular ATP, protects mice from lethal endotoxemia [53].
References
- Skulachev, V.P. Phenoptosis: Programmed death of an organism. Biochemistry 1999, 64, 1418–1426.
- Skulachev, V.P. Aging is a specific biological function rather than the result of a disorder in complex living systems: Biochemical evidence in support of Weismann’s hypothesis. Biochemistry 1997, 62, 1191–1195.
- Hamilton, W.D. The genetical evolution of social behaviour. I. J. Theor. Biol. 1964, 7, 17–52.
- Skulachev, V.P.; Shilovsky, G.A.; Putyatina, T.S.; Popov, N.A.; Markov, A.V.; Skulachev, M.V.; Sadovnichii, V.A. Perspectives of Homo sapiens lifespan extension: Focus on external or internal resources. Aging 2020, 12, 5566–5584.
- Jiang, W.; Wei, Y.; Long, Y.; Owen, A.; Wang, B.; Wu, X.; Luo, S.; Dang, Y.; Ma, D.K. A genetic program mediates cold-warming response and promotes stress-induced phenoptosis in C. elegans. eLife 2018, 7, e35037.
- Lohr, J.N.; Galimov, E.R.; Gems, D. Does senescence promote fitness in Caenorhabditis elegans by causing death? Ageing Res. Rev. 2019, 50, 58–71.
- Galimov, E.R.; Gems, D. Shorter life and reduced fecundity can increase colony fitness in virtual Caenorhabditis elegans. Aging Cell 2020, 19, e13141.
- Lidsky, P.V.; Andino, R. Epidemics as an adaptive driving force determining lifespan setpoints. Proc. Natl. Acad. Sci. USA 2020, 117, 17937–17948.
- Nirmala, J.G.; Lopus, M. Cell death mechanisms in eukaryotes. Cell Biol. Toxicol. 2020, 36, 145–164.
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54 Pt 1, 1–13.
- Barton, G.M.; Medzhitov, R. Toll-like receptors and their ligands. Curr. Top. Microbiol. Immunol. 2002, 270, 81–92.
- Janeway, C.A.; Medzhitov, R. Lipoproteins take their toll on the host. Curr. Biol. 1999, 9, R879–R882.
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820.
- Palm, N.W.; Medzhitov, R. Pattern recognition receptors and control of adaptive immunity. Immunol. Rev. 2009, 227, 221–233.
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384.
- Geijtenbeek, T.B.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479.
- Zhao, Y.; Karijolich, J. Know thyself: RIG-I-like receptor sensing of DNA virus infection. J. Virol. 2019, 93, e01085-19.
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128.
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21.
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14.
- Briard, B.; Place, D.E.; Kanneganti, T.D. DNA sensing in the innate immune response. Physiology 2020, 35, 112–124.
- Zhang, X.; Bai, X.C.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53.
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045.
- Seong, S.Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478.
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56.
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112.
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75.
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218.
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018, 38, 40–48.
- Aziz, M.; Brenner, M.; Wang, P. Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 2019, 106, 133–146.
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial cell-derived cytokines: More than just signaling the alarm. J. Clin. Investig. 2019, 129, 1441–1451.
- Ekaney, M.L.; Otto, G.P.; Sossdorf, M.; Sponholz, C.; Boehringer, M.; Loesche, W.; Rittirsch, D.; Wilharm, A.; Kurzai, O.; Bauer, M.; et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit. Care 2014, 18, 543.
- Quintana, F.J.; Cohen, I.R. Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J. Immunol. 2005, 175, 2777–2782.
- Ahrens, S.; Zelenay, S.; Sancho, D.; Hanc, P.; Kjaer, S.; Feest, C.; Fletcher, G.; Durkin, C.; Postigo, A.; Skehel, M.; et al. F-actin Is an evolutionarily conserved damage-sssociated molecular pattern Rrecognized by DNGR-1, a receptor for dead cells. Immunity 2012, 36, 635–645.
- Bukrinsky, M. Extracellular cyclophilins in health and disease. Biochim. Biophys. Acta 2015, 1850, 2087–2095.
- Riddell, J.R.; Wang, X.Y.; Minderman, H.; Gollnick, S.O. Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J. Immunol. 2010, 184, 1022–1030.
- Bozza, M.T.; Jeney, V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front. Immunol. 2020, 11, 1323.
- Calvo-Rodriguez, M.; García-Rodríguez, C.; Villalobos, C.; Núñez, L. Role of Toll like receptor 4 in Alzheimer’s disease. Front. Immunol. 2020, 11, 1588.
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317.
- Rodríguez-Nuevo, A.; Zorzano, A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019, 3, 195–207.
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323.
- Wenzel, T.J.; Bajwa, E.; Klegeris, A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129400.
- Tyurina, Y.Y.; Poloyac, S.M.; Tyurin, V.A.; Kapralov, A.A.; Jiang, J.; Anthonymuthu, T.S.; Kapralova, V.I.; Vikulina, A.S.; Jung, M.-Y.; Epperly, M.W. A mitochondrial pathway for biosynthesis of lipid mediators. Nat. Chem. 2014, 6, 542–552.
- Rocha, D.; Caldas, A.; Oliveira, L.; Bressan, J.; Hermsdorff, H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215.
- Fruitwala, S.; El-Naccache, D.W.; Chang, T.L. Multifaceted immune functions of human defensins and underlying mechanisms. Semin. Cell Dev. Biol. 2019, 88, 163–172.
- Yang, D.; Chen, Q.; Su, S.B.; Zhang, P.; Kurosaka, K.; Caspi, R.R.; Michalek, S.M.; Rosenberg, H.F.; Zhang, N.; Oppenheim, J.J. Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2-MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J. Exp. Med. 2008, 205, 79–90.
- Tewary, P.; Yang, D.; de la Rosa, G.; Li, Y.; Finn, M.W.; Krensky, A.M.; Clayberger, C.; Oppenheim, J.J. Granulysin activates antigen-presenting cells through TLR4 and acts as an immune alarmin. Blood 2010, 116, 3465–3474.
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001, 14, 303–313.
- Zhou, B.; Liu, J.; Zeng, L.; Zhu, S.; Wang, H.; Billiar, T.R.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Jiang, J.; et al. Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signalling. Nat. Microbiol. 2020, 5, 1576–1587.
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J. Histochem. Cytochem. 2018, 66, 213–227.
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337.
- Joosten, L.A.B.; Crişan, T.O.; Bjornstad, P.; Johnson, R.J. Asymptomatic hyperuricaemia: A silent activator of the innate immune system. Nat. Rev. Rheumatol. 2020, 16, 75–86.
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in sepsis. Front. Immunol. 2019, 10, 2536.
- Migeotte, I.; Communi, D.; Parmentier, M. Formyl peptide receptors: A promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006, 17, 501–519.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414.
- Orozco, L.D.; Bennett, B.J.; Farber, C.R.; Ghazalpour, A.; Pan, C.; Che, N.; Wen, P.; Qi, H.X.; Mutukulu, A.; Siemers, N.; et al. Unraveling inflammatory responses using systems genetics and gene-environment interactions in macrophages. Cell 2012, 151, 658–670.
- Sommer, S.S. Does cancer kill the individual and save the species? Hum. Mutat. 1994, 3, 166–169.
- He, Q.; Fu, Y.; Tian, D.; Yan, W. The contrasting roles of inflammasomes in cancer. Am. J. Cancer Res. 2018, 8, 566–583.
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941.
- McCall, K.D.; Muccioli, M.; Benencia, F. Toll-Like Receptors Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1223, 81–97.
- Wattenberg, M.M.; Beatty, G.L. Overcoming immunotherapeutic resistance by targeting the cancer inflammation cycle. Semin. Cancer Biol. 2020, 65, 38–50.
- Dib, P.R.B.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Andrade, F.B.; Hottz, E.D. Innate immune receptors in platelets and platelet-leukocyte interactions. J. Leukoc. Biol. 2020, 108, 1157–1182.
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb Thrombolysis 2021, 51, 446–453.
- Bonaventura, A.; Vecchie, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329.
- Bakeeva, L.E.; Barskov, I.V.; Egorov, M.V.; Isaev, N.K.; Kapelko, V.I.; Kazachenko, A.V.; Kirpatovsky, V.I.; Kozlovsky, S.V.; Lakomkin, V.L.; Levina, S.B.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke). Biochemistry 2008, 73, 1288–1299.
- Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Manskikh, V.N.; Pulkova, N.V.; Galkina, S.I.; Skulachev, V.P.; Zorov, D.B. Protective effect of mitochondria-targeted antioxidants in an acute bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, E3100–E3108.
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial damage and mitochondria-targeted antioxidant protection in LPS-induced acute kidney injury. Antioxidants 2019, 8, 176.
- Zakharova, V.V.; Pletjushkina, O.Y.; Galkin, I.I.; Zinovkin, R.A.; Chernyak, B.V.; Krysko, D.V.; Bachert, C.; Krysko, O.; Skulachev, V.P.; Popova, E.N. Low concentration of uncouplers of oxidative phosphorylation decreases the TNF-induced endothelial permeability and lethality in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 968–977.
- Asher, A.; Tintle, N.L.; Myers, M.; Lockshon, L.; Bacareza, H.; Harris, W.S. Blood omega-3 fatty acids and death from COVID-19: A pilot study. Prostaglandins Leukot Essent Fatty Acids 2021, 166, 102250.
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannata, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Inflamm. Res. 2021, 70, 7–10.
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma Emerg. Surg. 2020, 46, 751–775.
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999.
- Guo, Q.; Zhao, Y.; Li, J.; Liu, J.; Yang, X.; Guo, X.; Kuang, M.; Xia, H.; Zhang, Z.; Cao, L.; et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe 2021, 29, 222.e4–235.e4.
- Xue, J.; Suarez, J.S.; Minaai, M.; Li, S.; Gaudino, G.; Pass, H.I.; Carbone, M.; Yang, H. HMGB1 as a therapeutic target in disease. J. Cell Physiol. 2021, 236, 3406–3419.
- Xu, Z.; Huang, Y.; Mao, P.; Zhang, J.; Li, Y. Sepsis and ARDS: The dark side of histones. Mediators Inflamm. 2015, 2015, 205054.
- Palm, N.W.; Medzhitov, R. Not so fast: Adaptive suppression of innate immunity. Nat. Med. 2007, 13, 1142–1144.
- Kim, K.D.; Zhao, J.; Auh, S.; Yang, X.; Du, P.; Tang, H.; Fu, Y.-X. Adaptive immune cells temper initial innate responses. Nat. Med. 2007, 13, 1248–1252.
More
Information
Subjects:
Health Care Sciences & Services; Allergy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
COVID-19
Revisions:
3 times
(View History)
Update Date:
22 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No