Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Konstantin Lyamzaev and Version 3 by Bruce Ren.
Феноптоз, опосредованный врожденным иммунитетом, как частая причина смертности человека
Over-activation of innate immunity in response to bacterial or viral (including COVID-19) infections, massive trauma, or ischemia/reperfusion often causes severe illness and death. We suggest that such severe outcomes may be manifestations of an altruistic suicidal strategy protecting the entire population from the spread of pathogens and from dangerous pathologies rather than aberrant hyperstimulation of defense responses.
- pathogen-associated molecular patterns (PAMPs)
- damage-associated molecular patterns (DAMPs)
- mitochondrially-targeted antioxidants
- inflammation
- inflammasome
- programmed death
- phenoptosis
- COVID-19
1. Introduction
The concept of altruistic programmed death of whole organisms was proposed by Vladimir Skulachev more than two decades ago [1][2][1,2]. It was based on a suggestion that the death of individuals, if programmed, may be a subject to Darwinian selection and contribute to inclusive fitness. This suggestion expanded the original idea of inclusive fitness, also known as kin selection, which initially was about promoting reproduction and survival of genetic relatives only via social behavior [3]. In addition to this concept, Skulachev suggested that Darwinian selection may have shaped the mechanisms “of clearing a kin community of organisms or a population from individuals who have become unwholesome for this community”. He suggested to call this type of kin selection “phenoptosis”. Skulachev suggested that “the septic shock and stress-induced ischemic diseases of brain and heart” could be considered as the examples of phenoptosis [1]. At the same time, “slow phenoptosis” was proposed as an equivalent term for programmed aging [1][2][4][1,2,4].
The concept of phenoptosis has received support in experimental [5] and computer studies of Caenorhabditis elegans [6][7][6,7]. A cold shock followed by rewarming induced expression of genes of proteases, which promoted the programmed organismic death [5]. In the model of a clonal population of C. elegans that subsists on spatially limited food sources, simulations have shown that shorter lifespan can increase the colony fitness [6][7][6,7]. Presumably, phenoptosis, by reducing the futile food consumption by elderly or stressed worms leads to adaptive benefits for the colony. This concept is also fully consistent with the recently published results of mathematical modeling [8]. The authors demonstrated that in a population of short-lived individuals, the spread of infection is limited, and the pathogen clearance is more efficient than in population in which members live longer.
For diverse types of programmed cell death (PCD), such as apoptosis, necroptosis, and pyroptosis, specific execution mechanisms were identified [9]. Therefore, to categorize phenoptosis as a specific bona fide program, a specific biochemical execution mechanism(s) has to be identified as well.
2. Innate Immunity, Pathogen-Associated Molecular Patterns, and Pattern Recognition Receptors
The basic principle of innate immunity, as formulated by C. A. Janeway Jr [10], states that diverse invading pathogens are immediately recognized by molecules that are broadly shared by pathogens, such as glycans, N-formylated peptides, flagellin, and nucleic acids, but are distinguishable from host molecules. These molecules are collectively referred to as pathogen-associated molecular patterns (PAMPs). Some PAMPs are shared by viruses and diverse groups of microorganisms, including non-pathogenic ones.
PAMPs are recognized by pattern recognition receptors (PRRs). The first of the discovered PRRs are Toll-like receptors (TLR), of which there are at least 10 in humans [11][12][11,12]. Later several further classes of PRRs were discovered including C-type lectin receptors (CLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors, NOD (nucleotide-binding oligomerization domain)-like receptors (NLRs), and cytosolic DNA sensors [13][14][13,14].
TLRs are located in the plasma membrane or in the membranes of intracellular compartments (endoplasmic reticulum, endosomes, lysosomes), recognizing a wide variety of substances of microbial and viral origin [11][15][11,15].
CLRs make a large group of transmembrane or soluble receptors expressed primarily by myeloid cells. They recognize carbohydrates (glycans) on the surface of bacteria, fungi, helminths, and viruses [16].
The RIG-I-like receptors are intracellular viral RNA receptors. This family includes RIG-I that recognizes RIG-I circular RNA and short (≤300 bp) dsRNAs, MDA5 that senses long dsRNAs, and LGP2 that binds dsRNA and regulates signaling by RIG-I and MDA5 [17].
The NLR family has 22 members in humans, which are cytosolic sensors expressed in various immune and non-immune cells [18]. The NOD1 and NOD2 receptors recognize the degradation products of bacterial peptidoglycan, in particular muramyl dipeptide, which is characteristic of numerous pathogenic bacteria. The NLRP1, NLRP3, and NLRC4 receptors, in response to pro-inflammatory stimuli, form inflammasomes, large multiprotein complexes in which caspase-1 is activated to convert the precursors of interleukin 1β (pro-IL-1β) and interleukin 18 (pro-IL-18) into their active forms and to stimulate their release [18][19][20][18,19,20]. In addition to NLRs, the inflammasome formation can be induced by the interferon-inducible protein AIM2, the cytosolic DNA sensor [21].
The main PRR that recognizes the cytosolic DNA is the cyclic guanosine monophosphate adenosine monophosphate synthase (cGAS) that, by using STING (cGAS stimulator of interferon genes), stimulates the production of type-I interferon (IFN-I) [22]. Several other soluble receptors for cytosolic DNA and RNA have recently been identified [21]. All these sensors recognize DNA that can emerge during the life cycle of intracellular pathogens including viruses, bacteria, and parasites.
3. Damage Associated Molecular Patterns (DAMPs) and Their Receptors
In addition to PAMPs, the same TLRs, as well as some other PRRs, recognize damage-associated molecular patterns (DAMPs), which are endogenous molecules released from damaged cells, see [23][24][23,24] and Figure 1. Various types of DAMPs and their role in inflammation were extensively reviewed [25][26][25,26].
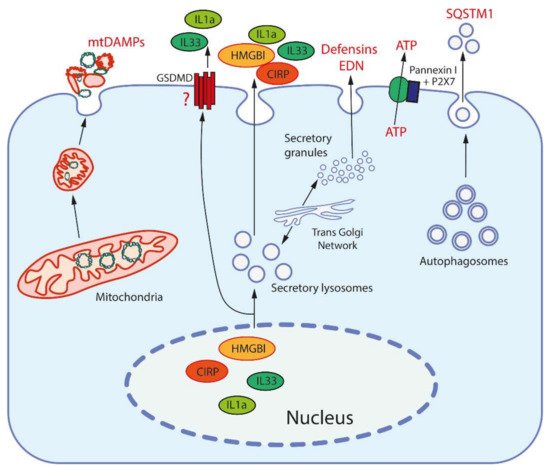
Figure 1. Release mechanisms for some DAMPs. High-mobility group protein B1 (HMGB1) and cold-inducible RNA-binding protein (CIRP) are released from the nucleus after modification to be entrapped and secreted by secretory lysosomes (which are produced by the trans-Golgi network). Interleukins 1a and 33 (IL1a, IL33) are also secreted by secretory lysosomes, but may be also released through the pores formed by gasdermin D (GSDMD). Mitochondria, after fragmentation, can be extruded from cells by an unknown mechanism(s). Secretory granules, as produced by the trans-Golgi network, release defensins and eosinophil-derived neurotoxin (EDN) by exocytosis. ATP is released through the pore formed by pannexin I complexed with the purinergic receptor P2X7. Sequestosome-1 (SQSTM1/h62) is an autophagosome receptor that can be released during secretory autophagy.
The concept of DAMPs stems from the “danger model” of immunity by P. Matzinger [23]. In contrast to the traditional “self versus non-self” recognition principle, she proposed that the immune system recognizes danger signals regardless of their origin. Initially, this hypothesis concerned adaptive immunity [23], but later the same principle was applied to innate immunity [24].
DAMPs can directly initiate at least two forms of PCD (pyroptosis and NETosis), which are involved in the pathogenesis of sepsis, as well as many other diseases associated with aseptic inflammation. Pyroptosis is a form of cell death that is accompanied by activation of a pore-forming protein gasdermin D by inflammatory caspases [27]. NETosis (where NET is deciphered as neutrophil extracellular traps) is a specific form of cell death that is characterized by the release of weblike DNA structures decorated with histones and antimicrobial proteins by neutrophils [28]. Cytokines produced in response to DAMP can also initiate other forms of PCD, such as apoptosis and necroptosis, which also contribute to various pathologies.
Today, a large number of DAMPs of various chemical nature and origins have been identified (see Table 1), and this list is expanding rapidly. The most important and well characterized DAMPs, namely the high-mobility group protein B1 (HMGB1) and cold-inducible RNA-binding protein (CIRP), function as chaperons in the nucleus [29][30][29,30]. Two interleukins of the IL-1 family, IL-1α and IL-33, are nuclear proteins capable of regulating expression. Outside the cell, they are recognized not by PRRs, but by the receptors of the IL1R family (IL-R1 and ST2, respectively), which transmit signals using the same MyD88 adapter as some TLRs [31]. Some other cytokines, such as thymic stromal lymphopoietin (TSLP) and IL-25, are also considered DAMPs although their intracellular functions have not been defined [31]. Expression of all these DAMPs is stimulated by various stresses and by mediators of inflammation.
Table 1. Typical damage-associated molecular patterns (DAMPs).
| # | Origin | DAMPs | Receptors | Ref | |
|---|---|---|---|---|---|
| 1 | Nucleus | HMGB1 | TLR2, TLR4, RAGE | [29] | |
| CIRP | TLR4, TREM-1 | [30] | |||
| Histones | TLR2, TLR4 | [32] | [47] | ||
| SAP130 | Mincle | [26] | |||
| IL-1α | IL1R1 | [31] | |||
| IL-33 | ST2 | [31] | |||
| DNA | cGAS, AIM2, RAGE, IFI16 | [21] | |||
| 2 | Cytosol | S100 proteins | TLR2, TLR4, RAGE | [31] | |
| HSPs | TLR2, TLR4, CD91 | [33] | [35] | ||
| F-Actin | DNGR-1, TREM1 | [34] | [32] | ||
| Cyclophilin A | CD147 | [35] | [33] | ||
| Peroxiredoxin 1 | TLR4 | [36] | [34] | ||
| Oxidized hemoglobin, heme | TLR4 | [37] | [36] | ||
| Amyloid β | TLR4 | [38] | [48] | ||
| ATP, ADP | P2X7R, P2Y2R, P2Y12R, | [39] | [40] | ||
| Uric acid | TREM-1, TLR2, TLR4, P2X7, NLRP3 | [26] | |||
| mRNA | TLR3 | [26] | |||
| microRNAs | TLR7 | [26] | |||
| SNAPIN | TLR2 | [26] | |||
| AGEs | RAGE | [26] | |||
| 3 | Mitochondria | Formyl peptides | FPR1 | [40] | [43] |
| mtDNA | TLR9, NLRP3 | [40] | [43] | ||
| Cardiolipin | NLRP3, TREM2 | [41] | [49] | ||
| Cytochrome | c | TLR4 | [42] | [44] | |
| Oxygenated mitochondrial fatty acids | TRL4 | [43][44] | [50,51] | ||
| TFAM | RAGE | [40] | [43] | ||
| 4 | ER, secretory granules, autophagosomes | Defensins | TLR4 | [45] | [37] |
| Cathelicidins | P2X7, FPR2 | [45] | [37] | ||
| Eosinophil-derived neurotoxin | TLR2 | [46] | [38] | ||
| Granulisin | TLR4 | [47] | [39] | ||
| Calreticulin | CD91 | [48] | [52] | ||
| Gp96 | TLR2, TLR4, CD91 | [48] | [52] | ||
| Sequestosome-1 (SQSTM1 or p62) | INSR | [49] | [53] | ||
| 5 | Extracellular matrix | Heparan sulphate, versican, aggrecan | TLR4 | [50] | [42] |
| Proteoglycans (biglycan, decorin, etc.) | TLR2, TLR4, CD14, NLRP3 | [50] | [42] | ||
| Tenascin-C | TLR4 | [50] | [42] | ||
| Fibrinogen | TLR4 | [50] | [42] | ||
| Fibronectin | TLR2, TLR4 | [50] | [42] | ||
| Low molecular weight hyaluronan | TLR2, TLR4, NLRP3 | [50] | [42] | ||
| 6 | Tumor cells | Annexin A1 | FPR1 | [51] | [45] |
| PAUF | TLR4 | [51] | [45] | ||
| API5 | TLR4 | [51] | [45] | ||
| Rps-3 | TLR4 | [51] | [45] |
Cytosolic DAMPs, such as S100 proteins that regulate Ca2+ signaling [31], actin, a component of cytoskeleton [34][32], peptidyl prolyl isomerase cyclophilin A [35][33], antioxidant protein peroxiredoxin 1 [36][34], and heat shock proteins (HSPs) [33][35] are among the most abundant proteins. Oxidized hemoglobin and heme, as released from red blood cells upon hemolysis, also act as DAMPs [37][36].
Secretory granules are another important source of DAMPs; these are antimicrobial peptides (defensins and LL37) [45][37], eosinophil-derived neurotoxin (EDN) [46][38], and granulisin, which accumulates in the granules of human cytotoxic T lymphocytes and natural killer cells [47][39].
Molecules of ATP [39][40] and uric acid (UA) [52][41] are the most important low molecular weight metabolites involved in the activation of inflammation upon release from the cell. In addition, various components of the extracellular matrix (ECM), including proteoglycans, soluble glycoproteins, low molecular weight hyaluronan, and heparan sulfate, are capable of acting as DAMPs [50][40][42,43].
Mitochondria are regarded as an important source of DAMPs (mtDAMPs) primarily due to their bacterial origin. Mitochondria-synthesized proteins are N-formylated, and mitochondrial DNA (mtDNA) contains insufficiently methylated CpG regions, similarly to the corresponding bacterial components which are recognized as PAMPs. One example of mtDAMPs in common with some bacteria is cytochrome c [42][44]. In contrast, the transcription factor A (TFAM) is an example of a mtDAMPs that appears not to have a bacterial counterpart [40][43]. Almost the entire list of known DAMPs can be released from tumors and contribute to immunogenic cell death (ICD) associated with an adaptive immune response [51][45]. Some of the DAMPs (not all), including those that are predominantly released from tumors, are listed in Table 1.
DAMPs can be released from cells via active and passive pathways (Figure 1). Passive release requires the rupture of the plasma membrane and occurs during pyroptosis, necroptosis, or necrosis. Both nuclear DNA and histones can be released from leucocytes in form of decondensed chromatin forming extracellular traps (ETs; NETs in the case of neutrophils). Interestingly, some DAMPs (HMGB1, CIRP) can induce release of NETs, which leads to the formation of an amplification loop [53][46].
Active release of nuclear DAMPs, such as HMGB1 and CIRP, requires their posttranslational modification in order to leave the nucleus, and subsequently occurs through exocytosis of secretory lysosomes [29][30][29,30]. The same exocytotic mechanism participates in active release of cytosolic and mitochondrial DAMPs. ATP can be released not only from damaged cells, but also through the channel formed by the P2X purinoreceptor 7 (P2X7 receptor) together with pannexins [39][40]. Uric acid excretion is largely regulated by urate transporters in the renal tubules [52][41]. In general, it can be assumed that the release of the most important DAMPs may be mediated by specific mechanisms in living cells (Figure 1).
Initially, it was thought that DAMPs are only recognized by pattern recognition receptors (PPRs), but later the receptors for advanced glycation end products (RAGE), triggering receptor expressed on myeloid cells 1 (TREM-1), CD91 and purinergic receptors were also shown to participate in the DAMP recognition [25][26][25,26]. It has recently been reported that sequestosome 1 (SQSTM1/p62), a known selective autophagy receptor released by macrophages and monocytes, is recognized by the insulin receptor (INSR) to stimulate inflammation [49][53]. In addition, formyl peptide receptor 1 (FPR1), is a G protein-coupled receptor that recognizes N-formylmethionine-containing peptides [54].
Some DAMPs can be recognized without release from the cell. MtDNA, released from mitochondria into cytosol and oxidized, can stimulate the NLRP3 inflammasome activity [55]. Another intracellular DAMP is cardiolipin, a four-tail phospholipid which is located exclusively in the inner mitochondrial membrane. Under stress, cardiolipin is externalized on the mitochondrial surface and stimulates the assembly of the NLRP3 inflammasome [41][49].
Signal transduction from DAMP-sensing receptors uses some of signaling modules that are activated by elevated cytosolic levels of Ca2+ and reactive oxygen species (ROS). The main outcomes of DAMP-dependent signaling include NFκB and inflammasomes activation, followed by the production of inflammatory cytokines and chemokines, activation of mitogen-activated protein kinases (MAPK), and stimulation of interferon signaling (Figure 2), see [26] for a comprehensive review.
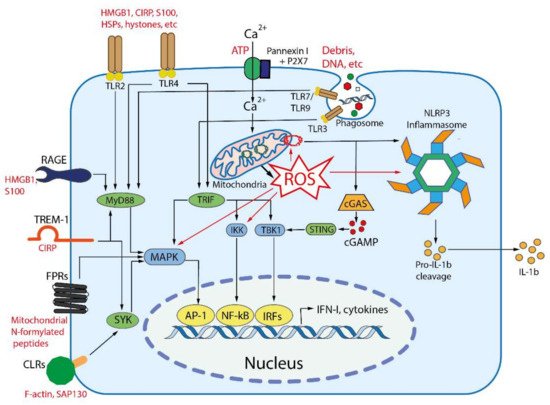
Figure 2. Main DAMP-sensing receptors and signaling. DAMPs can be recognized by membrane receptors, such as Toll- like receptors (TLRs), receptor for advanced glycation end products (RAGE), triggering receptor expressed on myeloid cells 1 (TREM-1), formyl peptide receptors (FPRs), C-type lectin receptors (CLRs), purinergic receptor P2X7, as well as by cytoplasmic receptors such as cyclic GMP-AMP synthase (cGAS) and NLR family pyrin domain containing 3 (NLRP3) inflammasome. Endogenous nuclear and mitochondrial DNA released into the cytoplasm activate cGAS, which produces cyclic GMP-AMP (cGAMP) that binds to stimulator of interferon genes (STING). Activated STING stimulates the kinases TBK1 and IKK, leading to the expression of type-I interferons (IFN-I) and inflammatory cytokines. TLR2 and TLR4 can be activated by a variety of extracellular DAMPs, whereas TLRs 3, 7, and 9 are activated by phagocytosed nucleic acids in endosomes. TLR-dependent signaling is mediated by the adaptor proteins: myeloid differentiation primary-response 88 (MyD88), and TIR-domain-containing adaptor inducing IFNβ (TRIF). The both adaptors activate mitogen-activated protein kinases (MAPKs) and IκB kinase (IKK), leading to activation of the transcription factors activator protein 1 (AP-1) and nuclear factor κB (NFκB), which stimulate the expression of inflammatory cytokines. TRIF also activates TANK binding kinase 1 (TBK1), which activates the interferon regulatory factors (IRFs) leading to the expression of type I interferon (IFN-I). RAGEs, which recognize HMGB1, S100 and some other DAMPs, also activate MyD88. TREM-1, which recognizes CIRP, activates spleen tyrosine kinase (SYK), that further stimulates MyD88, MAPKs and NFκB. Several C-type lectin receptors (CLRs) recognize F-actin and SAP130 and also activate SYK. Formyl peptide receptors (FPRs) are G-protein coupled receptors that recognize mitochondrial N-formylated peptides and stimulate several signaling pathways (including MAPKs) to activate neutrophils. P2X7 is ionotropic receptor that opens Ca2+ channel in response to extracellular ATP. An increase in the concentration of Ca2+ in cytoplasm modulates various signaling pathways and stimulates the production of reactive oxygen species (ROS) in mitochondria. Elevated ROS levels are critical for the activation of MAPKs, NFκB and the NLRP3 inflammasome. ROS-oxidized mitochondrial DNA is recognized by NLRP3.
In sum, DAMPs, as well as PAMPs, initiate an innate immune response, which involves the release of cytokines and other inflammatory mediators, activation of some specific defense mechanisms (for example, the production of extracellular traps), and induction of certain types of programmed cell death. Such a combination of organismic reactions is commonly called inflammation, which has been recently defined as “the innate immune response to harmful stimuli such as pathogens, injury, and tissue malfunction” [56]. This definition covers both acute (local or systemic) and chronic inflammatory processes.