In December 2019, several cases of pneumonia of unknown etiology were reported in Wuhan, China. The outbreak, which was reported to have commenced in late December 2019 in China, was soon worldwide, with increased cases and deaths
[1]. The causative agent of the outbreak earned the name SARS-CoV-2 after being identified as a beta coronavirus whose genomic sequence was closely aligned to severe acute respiratory syndrome coronavirus (SARS-CoV), earlier identified in 2003
[1][2][3]. SARS-CoV-2 has been identified as the seventh coronavirus known to infect humans
[4]; SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2 have been reported to be severe, while human coronavirus (HCov)-HKU1, HCov-NL63, HCoV-OC43, and HCoV-229E are mild
[5]. As of 5:52 pm CEST, 11 October 2021, the WHO had identified 237,383,711 confirmed cases of COVID-19, including 4,842,716 deaths globally
[6].
SARS-CoV-2, including the recently reported severe variant B.1.617.2
[7][8], attacks the respiratory tract with symptoms ranging from breathing difficulties, sore throat, high fever, diarrhea, and cough to multiple organ failure and ultimately death
[9]. This can occur when the spike protein (Spro), which possesses an S1 domain and an S2 subunit, binds to angiotensin-converting enzyme-2 (ACE-2) receptors on the surface of the host alveoli to allow for entry
[10]. Thus, studies have identified the Spro as a drug target to prevent interaction with ACE-2, thereby inhibiting the entry of the virus
[11][12]. Furthermore, with the aid of RNA-dependent RNA polymerase (RdRp), the viral RNA translation results in the synthesis of proteins responsible for synthesizing new virions from single-stranded RNA, which makes RdRp a primary drug target to prevent viral growth and replication
[13][14]. Overall, viral studies have shown that viral proteases are typical targets for anti-viral drug development
[15][16]. Hence, the main protease (Mpro) of SARS-CoV-2, which plays a role in viral replication, could be a potential therapeutic target
[17][18].
Heterocyclic compounds play an essential role in drug discovery and development; hence, great work has gone into developing simple and environmentally friendly methods for their high yielding production
[21]. For example, compounds derived from azetidine have shown to have a diverse range of pharmacological activities, such as anticancer
[22], antibacterial
[23][24], antimicrobial
[25], antischizophrenic
[26], antimalarial
[27][28], antiobesity
[29][30], anti-viral
[31], antioxidant
[31], and dopamine antagonist
[32] activity, amongst others. They are also reported to be tRNA-synthetase inhibitors, signal transducers and activators of transcription-3 (STAT-3) inhibitors, and vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors
[31][33][34][35]. Other heterocyclics, such as cyclic amidine and guanidine also possess biological potency
[36][37]. Heterocyclic skeletons of promising pharmacological importance usually contain nitrogen, sulphur, and oxygen as they represent a major proportion of the bioactive heterocyclic compounds and marketed drugs
[21]. Azetidine, amidine, and gunanine rings are nitrogen-containing heterocyclic organic compounds with different synthetic strategies and pharmaceutical importance
[31][32][37][38]. As a result, pharmaceutical companies incorporate these compounds into the design, formulation, development, and synthesis of drugs. Specifically, the antiproliferative properties of azetidine derivatives have prompted their use and production in anti-viral research
[31][39][40]. Their anti-viral activities are aided by their ability to inhibit proteins essential to the viral life cycle
[41][42]. Some of these heterocyclic drugs with anti-viral activities have been approved by the FDA. In 2018, the FDA approved baricitinib, a heterocyclic derivative of azeitidine, to treat patients with rheumatoid arthritis
[31]. Recently, baricitinib was found to engender early stabilization of the respiratory functions and reduce rehospitalization and the mortality rate resulting from COVID-19 at a daily high dose
[43]. Barticinib combined with other drugs has been found to control the virus. Treatment of the virus with baricitinib plus hydroxychloroquine was associated with recovery in 11 of 15 patients
[44]. Treatment with baricitinib plus dexamethasone resulted in the reduced mortality of COVID-19 drugs
[45]. Moreover, Baricitinib plus remdesivir was reported to be superior to remdesivir alone as it reduces recovery time and accelerates improvement in clinical status among patients with COVID-19, notably among those receiving high-flow oxygen or noninvasive ventilation
[46].
2. Current Insights
As a result of the increased cases and mortality rates resulting from the global health challenge, the coronavirus, there have been responses to the emergency call to develop mitigation strategies for the infection. Some of the strategies being used to control the viral spread of the disease are the recommended use of face masks, hand gloves, and sanitizers. While quite a number of vaccines are being developed to stimulate the production of antibodies against the virus, there are still concurrent investigations exploring the therapeutic potential of some anti-viral agents, including remdesivir
[47][48]. As the search for new anti-SARS-CoV-2 drugs continues, the use of heterocyclic organic compounds in anti-viral drug development is essential as they have been reported to be efficacious against other major viral infections
[31][49][50]. Heterocyclic derivatives, such as guanine, cyclic amidine, and azetidine exhibit anti-viral activity as they inhibit the regulation of the enzymes and proteins associated with the life cycle of viruses
[31][51]. Some of these heterocyclic derivatives have been approved by the US Food and Drug Administration (FDA) for treatment of some diseases. In 2017, delafloxacin was approved to treat patients with acute bacterial skin infections
[31]. The FDA also approved the calcium channel blocker, azelnidipine, alone, or combined with other antihypertensive drugs, to treat hypertension
[31]. In 2018, the FDA approved baricitinib to treat patients with rheumatoid arthritis, and it has been found to stabilize the respiratory functions of COVID-19 patients at a daily high dose
[31][43].
The high binding affinities exhibited by the test compounds towards the SARS-CoV-2 target proteins in this study indicate the inhibitory potential of these compounds against these biomolecules and their possible roles as therapeutic agents against SARS-CoV-2. In addition, this study observed that C13 possesses a higher binding affinity for the Spro and RdRp target proteins than for the other compounds, while C15 has a higher binding affinity for Mpro. This could be linked to the presence of the delocalized п electrons in the aromatic ring and the presence of polar compounds in the structure of these compounds. Approximately 20% of essential amino acids are structurally aromatic; interactions involving aromatic compounds are essential to biological recognition, including protein–ligand interaction
[9]. Singh
[52] reported that aromatic compounds are crucial in drug design as they engender improved efficacy and lead to optimization of the drug. The inhibitory potential of these derivates is consistent with results from other studies. The treatment of COVID-19-hospitalized adults with baricitinib, as well as dexamethasone, resulted in reduced mortality arising from the compounds’ inhibitory potential
[45].
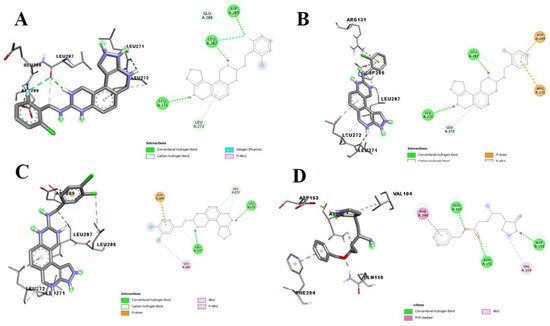
The molecular docking studies revealed the binding pose of the compounds with the highest docking scores compared to the standard ligands. Analysis of the 3D and 2D structures of the docked SARS-CoV-2 target test compound complex showed that these compounds possess inhibitory potentials against the proteins. Compounds C15, C12, and C14, interacted with LEU A:271, LEU A:287, ASP A:289, GLU A:288, and LEU A:272 at the binding pocket of the SARS-CoV-2 Mpro, while its co-crystallized ligand, N3, interacted with GLN A:110, ASP A:153, ASP A:151, VAL A:104, and PHE A:294 (
Figure 1). In this study, there was no interaction with CYS A: 145 and A: HIS 41, amino acids responsible for catalysis at the catalytic site of the main protease of SARS-CoV-2, as reported by Kneller et al.
[53]. Present in the protease’s catalytic dyads are other amino acids from CYS 145 and HIS 41, which are functional in eliminating Mpro enzymes. Dimerization and mutations of Mpro that engender enzymes with reduced activities are associated with interactions with the residues around GLU 288, ASP 289, and GLU 290
[54]. While CYS 145 and HIS 45 mediate the catalytic mechanism of the enzyme, mutation of these residues leads to the total annihilation of the Mpro activity
[55][56]. Hence, the interactions of these compounds with GLU 288 and ASP 289 amongst other amino acids suggest that they could interfere with the catalytic activity of Mpro, inhibit SARS-CoV-2 replication, and ultimately eliminate Mpro by mutation or dimerization.
Figure 1. 3D (left) and 2D (right) views of the molecular interactions of amino-acid residues of Mpro (6LU7) with (A) C15, (B) C12, (C) C14, and (D) N3.
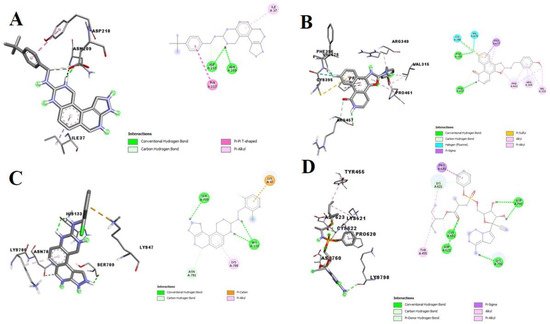
The SARS-CoV-2 Spro interacted with C13, C11, and C4 through critical amino acid residues, including the PHE A:486 of its receptor-binding domain (RBD) (
Figure 2). PHE 486 has been identified as one of the critical residues that bind Spro to the ACE2 receptor
[57]. PHE 486 of SARS-CoV-2 has similar biochemical properties to LEU 472 of SARS-CoV
[55]. In addition, PHE 486 of the spike receptor-binding motif, through hydrophobic interactions, binds with the GLN24, LEU79, MET82, and TYR83 of ACE2
[55][58]. Moreover, from this study, the compounds—C13, C11, and C4—interacted similarly with the HIS A:540 and PRO A:415 residues of Spro RBD. Being similar in their structures to PHE, the heterocyclic and unsaturated structures of HIS and PRO could be responsible for the interaction. Therefore, pharmacological interaction between the test compounds and the critical amino acid residue, PHE A:486, could limit the binding of SARS-CoV-2 Spro to the ACE2 receptor host alveoli, thereby limiting viral entry and circumventing the progression of the disease.
Figure 2. 3D (left) and 2D (right) views of the molecular interactions of amino-acid residues of Spro (6LZG) with (A) C13, (B) C11, (C) C4, and (D) remdesivir.
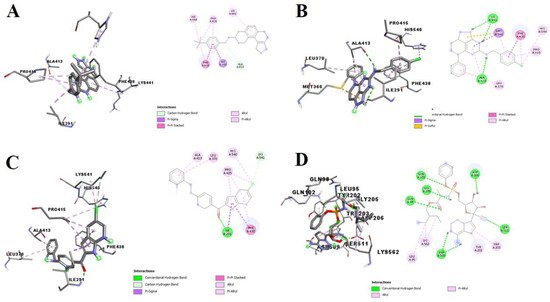
Furthermore, C2, C8, and C11 demonstrated to be strong potential inhibitors of RdRp. Unlike the amino acid residue interaction with remdesivir, these compounds interacted with different and unique amino acids in the binding pocket of SARS-CoV-2 RdRp (
Figure 3). In this study, the test compounds interacted through conventional hydrogen bonds, halogen bonds, carbon-hydrogen bonds, alkyl interactions, and п-alkyl interactions with amino acids—VAL A:675, PRO A:677, SER A:709, ASP A:216, ASN A:209, TYR A:217, ILE A:37, PHE A:396, ARG A:457, CYS A:395, PRO A:461, ARG A:349, VAL A:315, HIS A:133, LYS A:47, SER A:709, HIS A:133, LYS A:780, TYR A:455, PRO A:620 and VAL A:315—present in the binding pocket of SARS-CoV-2. Kumar
[59] similarly reported the amino acid residues—LYS47, ASN781, and SER709—in the binding pocket of SARS-CoV-2. Thus, Spro can potentially form hydrogen bonds with the drug molecule. In addition, RdRp plays an essential role in viral RNA translation, resulting in a protein that produces new virions from single-stranded RNA
[13][14]. Hence, its inhibition by these heterocyclic compounds could offer therapeutic benefits against the SARS-CoV-2 growth and replication.
Figure 3. 3D (left) and 2D (right) views of the molecular interactions of amino-acid residues of RdRp (6M71) with (A) C13, (B) C9, (C) C15, and (D) remdesivir.
The accuracy of the docking protocol was validated by re-docking some of the standard and test ligands at a minimized energy back into the binding pocket of the drug targets. As previously stated, the re-docked pose almost completely overlapped the experimental orientation, indicating that AutoDock Vina on PyRx re-docked the standard and test ligands back into the binding pocket of the target proteins with a high degree of accuracy and precision. This demonstrates that the docking methodology used in this study is reliable and that the docking scores obtained are correct. Moreover, Ambrose et al.
[60] validated this using a similar docking protocol by re-docking the co-crystallized ligand (PDB Ligand ID: 2WR) with the mutant EGFR (PDB: 3W2S) studied. It was evident in his study that there was a nearly perfect overlap of the re-docked ligands.
In addition to the inhibitory potentials demonstrated by the test compounds towards the drug targets, the compound possesses moderate ADMET properties. However, the compound may require lead optimization of its properties while maintaining its binding affinity. ADMET analysis is collectively known as absorption, distribution, metabolism, elimination, and toxicity. It is an analysis that determines whether a molecule can be easily absorbed, delivered to its target site of action, digested in a way that does not eliminate activity, and easily removed from the body while preventing toxic effects. A high-quality drug candidate should be effective against the therapeutic target and have appropriate ADMET properties at a therapeutic dose
[61]. As a result, many in silico models for predicting chemical ADMET properties have been created and it has become advantageous as it reveals a pharmacokinetics-related failure of drugs before proceeding to the clinical phase
[9].
Lipophilicity is generally considered a key determinant of permeability across tissue membranes, while water solubility is another physicochemical property that determines a drug’s ADMET behaviours
[9]. Orally administered drugs usually have a high lipophilic value, indicating easy passage and absorption through the intestinal lining, penetration of the membrane of the target cells, and travel in the blood. There is a direct relationship between the log P value and lipophilicity, but this negatively correlates with water solubility
[62]. Hence, the test compounds with Log P values between 3.27 and 4.10 (C4, C9, C11, C12, C13, C14, and C15) came out to be moderately soluble (
Table 1). The presence of unsaturated structures, polar solar chains, and the higher molecular weights of the azeditine derivatives may have contributed to the moderate solubility of these compounds.
Table 1. Predicted lipophilicity (Log P), water solubility (Log Sw), drug-likeness, and bioactivity of selected compounds and standard ligands.
| Parameters |
C4 |
C9 |
C11 |
C12 |
C13 |
C14 |
C15 |
N3 |
Remdesivir |
| Molecular weight (g/mol) |
380.8 |
389.38 |
376.84 |
362.82 |
384.48 |
397.26 |
380.81 |
290.36 |
602.58 |
| Consensus Log P |
3.98 |
4.1 |
3.63 |
3.27 |
3.93 |
3.78 |
3.55 |
1.48 |
1.56 |
| Log Sw (Silicos-IT) |
5.03 |
5 |
3.72 |
4.22 |
4.92 |
4.85 |
4.63 |
2.35 |
−0.05 |
| Solubility class |
Moderately soluble |
Moderately soluble |
Moderately soluble |
Moderately soluble |
Moderately soluble |
Moderately soluble |
Moderately soluble |
soluble |
Moderately soluble |
| #Heavy atoms |
27 |
29 |
27 |
26 |
29 |
27 |
27 |
21 |
42 |
| #Aromatic heavy atoms |
21 |
23 |
21 |
21 |
21 |
21 |
21 |
6 |
15 |
| Fraction Csp3 |
0.05 |
0.09 |
0.15 |
0.16 |
0.3 |
0.16 |
0.16 |
0.5 |
0.48 |
| #Rotatable bonds |
5 |
4 |
4 |
3 |
4 |
3 |
3 |
8 |
14 |
| #H-bond acceptors |
4 |
5 |
3 |
4 |
4 |
4 |
5 |
4 |
12 |
| #H-bond donors |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
4 |
| MR |
102.52 |
109.88 |
110.72 |
101.54 |
115.8 |
106.55 |
101.49 |
83.47 |
150.43 |
| TPSA (Å2) |
70.67 |
80.15 |
69.73 |
79.38 |
79.38 |
79.38 |
79.38 |
81.42 |
213.36 |
| Lipinski violations |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
2 |
| Ghose violations |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
3 |
| Veber violations |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
2 |
| Egan violations |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
| Muegge violations |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
3 |
| Bioavailability Score |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
0.17 |
| Synthetic availability |
2.75 |
3.07 |
3.13 |
3.04 |
3.45 |
3.07 |
3.11 |
2.9 |
6.33 |
Drug-likeness is established based on chemical structures and physicochemical properties and is a qualitative assessment of oral bioavailability
[63]. Moreover, Lipinski’s Rule states that for an orally active drug, the following conditions must be obeyed: ≤5 H-bond donors, ≤10 H-bond acceptors, a molecular weight ≤500 g/mol, and a log
p ˂ 5.43; a ligand is considered orally inactive if it violates two or more of Lipinski’s rules
[64]. Considering these criteria, all the selected compounds (C4, C9, C11, C12, C13, C14, and C15) meet the requirements for oral bioavailability (
Table 2). Moreover, none of the test compounds violated Veber’s rule, whose criteria are the presence of rotatable bonds ≤10 and polar surface (TPSA) area ≤140 Å2
[65]. Moreover, evident from the bioavailability score of 0.55%, all the selected test compounds will be good oral drugs (
Table 2). This result shows the drug-likeness of these ligands compared to the standard and co-crystallized ligands.
Table 2. Toxicity profile prediction of test compounds.
| Parameters |
C4 |
C9 |
C11 |
C12 |
C13 |
C14 |
C15 |
N3 |
Remdesivir |
| hERG-Blockers |
- |
- |
- |
- |
- |
- |
- |
- |
- |
| H-HT (Human Hepatotoxicity) |
- |
- |
- |
- |
- |
- |
- |
- |
- |
| AMES (Ames Mutagenicity) |
- |
- |
- |
- |
- |
- |
- |
- |
- |
| LD50 (LD50 of acute toxicity) |
2.503-log mol/kg (1195.937 mg/kg) |
2.616-log mol/kg (942.712 mg/kg) |
2.569-log mol/kg (1016.646 mg/kg) |
2.579-log mol/kg (956.524 mg/kg) |
2.704-log mol/kg (760.119 mg/kg) |
2.651-log mol/kg (887.329 mg/kg) |
2.598-log mol/kg (960.977 mg/kg) |
2.452-log mol/kg (1025.513 mg/kg) |
2.989-log mol/kg (618.042 mg/kg) |
| DILI (Drug Induced Liver Injury) |
- |
- |
- |
- |
- |
- |
- |
- |
- |
| FDAMDD (Maximum Recommended Daily Dose) |
- |
- |
- |
- |
- |
- |
- |
- |
- |
According to the pharmacokinetic predictions of the compounds, all the test compounds were predicted to be inhibitors of CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4, except for compound C12, which is not an inhibitor of CYP2C9 (
Table 3). Cytochrome P450 (CYP) is an isoenzyme superfamily that catalyzes various biochemical processes in phase I drug metabolism (Hollenberg, 2002). The inhibition of the five main isoforms—CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4—from eventually becoming the substrates of medications is a primary cause of pharmacokinetics-related drug-drug interactions
[62][66].
Table 3. Pharmacokinetics prediction output of test compounds.
| Parameters |
C4 |
C9 |
C11 |
C12 |
C13 |
C14 |
C15 |
N3 |
Remdesivir |
| GI Absorption |
High |
High |
High |
High |
High |
High |
High |
High |
Low |
| Blood-brain permeant |
Yes |
No |
Yes |
No |
No |
Yes |
No |
No |
No |
| Pgp substrate |
No |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
| CYP1A2 inhibitor |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
No |
No |
| CYP2C19 inhibitor |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
No |
No |
| CYP2C9 inhibitor |
Yes |
Yes |
Yes |
No |
Yes |
Yes |
Yes |
No |
No |
| CYP2D6 inhibitor |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
No |
No |
| CYP3A4 inhibitor |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
No |
Yes |
| Skin permeant Log Kp (cm/s) |
−5.46 |
−5.62 |
−5.27 |
−6.04 |
−5.42 |
−5.27 |
−6.07 |
−7.50 |
−8.62 |
All the selected test compounds are predicted not to be substrates of Pgp, except compound C4. Pgp is an ATP-binding cassette transporter responsible for the active efflux of xenobiotics across biological membranes to protect the body against foreign toxins and to contribute to drug resistance
[9]. This result infers that all the aforementioned compounds, aside from C4, are likely to be prevented from entering into their target sites of action due to the active efflux of the ATP-binding cassette. Nevertheless, the results of the toxicity prediction showed none of the compounds had tendencies towards any of the toxicity parameters tested. Therefore, the compounds could be considered for experimental studies and further development into novel drugs to treat SARS-CoV-2.
 +1 credit
+1 credit