Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gurevich Evgenia | + 2408 word(s) | 2408 | 2021-12-06 04:15:58 | | | |
| 2 | Gurevich Evgenia | Meta information modification | 2408 | 2021-12-16 15:31:56 | | | | |
| 3 | Camila Xu | + 286 word(s) | 2694 | 2021-12-17 01:42:21 | | | | |
| 4 | Camila Xu | + 286 word(s) | 2694 | 2021-12-17 01:43:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Evgenia, G. Growth Hormone and IGF1 Actions in Kidney. Encyclopedia. Available online: https://encyclopedia.pub/entry/17214 (accessed on 26 July 2026).
Evgenia G. Growth Hormone and IGF1 Actions in Kidney. Encyclopedia. Available at: https://encyclopedia.pub/entry/17214. Accessed July 26, 2026.
Evgenia, Gurevich. "Growth Hormone and IGF1 Actions in Kidney" Encyclopedia, https://encyclopedia.pub/entry/17214 (accessed July 26, 2026).
Evgenia, G. (2021, December 16). Growth Hormone and IGF1 Actions in Kidney. In Encyclopedia. https://encyclopedia.pub/entry/17214
Evgenia, Gurevich. "Growth Hormone and IGF1 Actions in Kidney." Encyclopedia. Web. 16 December, 2021.
Copy Citation
Growth hormone (GH) exerts multiple effects on different organs including the kidneys, either directly or via its main mediator, insulin-like-growth factor-1 (IGF-1).
growth hormone
insulin-like growth factor 1
growth hormone receptor
1. Introduction
Most animals must undergo a transition from maternal environment to independent life through processes of growth and maturation. Important hormonal regulators of childhood growth are growth hormone (GH), insulin-like growth factor 1 (IGF1), sex steroids, and thyroid hormone. GH and IGF1 are part of an axis, which is essential for bone and organs growth. The kidneys express both GH as well as IGF1 receptors, and are one of the key target organs for these hormones’ actions.
2. Normal GH-IGF1 Axis and Physiology
GH is produced by somatotroph cells of the anterior pituitary and secreted in a pulsatory way under the positive control of hypothalamic GH-releasing hormone (GHRH) and the negative control of somatostatin [1]. The response to GHRH is mediated via GH-releasing hormone receptor (GHRHR), a G protein–coupled receptor (GPCR) expressed specifically in somatotrophs [2]. Other factors such as insulin-like growth factor (IGF1), neuropeptide Y, and hyperglycemia inhibit GH secretion, and hypoglycemia, thyroxine, ghrelin, klotho, and glucocorticoids stimulate GH secretion [3].
GH acts by binding to GH receptor (GHR) to stimulate, among other genes, the synthesis of insulin-growth factor-1 (IGF1). The bioavailability of GH is regulated by GH-binding protein (GHBP), which is the extracellular part of GHR. Intracellular signal transduction after GH binding to its receptor requires the activation of Janus-associated kinase 2 (JAK2) [4], which stimulates phosphorylation of signal transducer and activator of transcription (STAT) proteins MAPK and PI3K. STAT proteins migrate to the nucleus, activating, among others, gene transcription of IGF1, the main mediator of GH action. In addition, suppressors of cytokine signaling (SOCS) are activated, which dephosphorylate STAT, leading to a negative feedback action on GH [5]. Circulating IGF1 suppresses pituitary GH secretion in a negative feedback loop. IGF1 is synthesized mostly in the liver, but also in peripheral tissues under GH regulation, although nutrition, insulin, thyroid, and sex hormones also affect its expression [6]. The effects of IGF1 are mediated by the type 1 IGF receptor (IGF1R) in a signaling pathway similar to insulin/insulin receptor (IR). IGF1R and IR share amino acid identity, and can be activated both by insulin, IGF1, and IGF2. [7]. IGF1R is a membrane-bound tyrosine kinase heterotetramer, and its activation leads to autophosphorylation of tyrosine residues, leading to signal transduction [8]. The bioactivity of circulating IGF1 is modulated by IGF-binding proteins (IGFBPs 1-6), which facilitate its stability in serum and extracellular matrices. Most IGFs in serum are bound to IGFBP and the acid-labile subunit (ALS), a protein that stabilizes IGF [9][10], and this complex serves as reservoir of IGFs, keeping serum concentration of free IGFs constant. Plasma concentration of IGFBP3 and ALS are also increased by GH, similar to IGF1.
3. GH-IGF1: Axis or Independent Functions?
Whereas GH is only synthesized in pituitary, GHR and IGF1 are expressed in many tissues including the kidneys. Originally GH action was thought to be mediated only through IGF1, called somatomedin, without any direct effects (“somatomedin theory”) [11]. Later, “dual effector hypothesis” suggested that GH also acts directly to promote cell differentiation, independent of IGF1 [12][13][14]. Concentrating on kidneys as one of the target organs for both GH and IGF-1, GH treatment increased kidney IGF1 mRNA levels in hypophysectomized rats, confirming local renal IGF1 production [15]. IGF1 levels are higher in renal venous blood than in renal arterial blood, suggesting significant renal IGF1 biosynthesis [16]. Evidence for a direct IGF1 action in the kidney also comes from studies showing that prolonged treatment with recombinant human (rh) IGF1 increased kidney size in hypophysectomized rats [17] and enhanced the glomerular filtration rate (GFR) in healthy men [18].
4. Observations from Knockout and Transgenic Animals
Animal models of gene inactivation, as well as pathophysiological models, provide important data on mechanisms and role of GH/IGF1 in renal organogenesis. Evidence of both dependent and independent functions of GH and IGF1 on the kidney come from genetically engineered animal models (see Table 1 and Table 2). Examples of the knockout models, where mutations were introduced in every step of the axis (GHRH → GH → GHR/GHBP → JAK2 → STAT5 → IGF1 → IGF1R), followed by the transgenic models, overexpressing genes along this axis, are discussed here.
Table 1. Chain of GH-IGF signals: general and kidney phenotypes with loss of function. KO: knockout muse model; NA: not available; m: mouse; h: human.
| KO/Human Mutation General Phenotype | KO/Kidney Phenotype | Ref. | |
|---|---|---|---|
| GH | Growth retardation | Disproportionally small kidneys | [17] |
| GHR/ GHBP |
Growth retardation after birth, low IGF1, greater longevity | Disproportionally small kidneys Protection against diabetic nephropathy |
[18] |
| JAK2 | Embryonic lethality due to a lack of hematopoiesis | NA | [19] |
| STAT5 | Abnormal postnatal growth, facial dysmorphism, immunodeficiency (h) perinatal death, dwarfism, anemia, immunodeficiency (m) |
NA | [20][21] |
| IGF1 | Severe growth retardation, infertility, deficiencies in bone and muscle development, lethal respiratory failure | Proportionally small kidneys, decreased glomerular size and nephron number Liver specific IGF1 KO mice: compensatory remnant kidney hypertrophy after unilateral nephrectomy, no significant change in IGF1R phosphorylation (despite markedly decreased kidney IGF-1 levels) | [22][23][24] |
| IGF1R | Respiratory failure, low birth weight, developmental abnormalities, perinatal death | NA | [25] |
| SOCS2 | Gigantism, improved somatic growth in CKD model | No glomerulosclerosis development | [26] |
| IGFBP1 | indistinguishable from wild-type, no embryonic lethality | NA | [27] |
| IGFBP2 | minor gender specific changes in bone structure, minor changes in the weights of spleen and liver in adult males | NA | [28][29] |
| IGFBP3 | Normal | NA | [30] |
| IGFBP4 | mild 10%–15% reduction in prenatal growth | NA | [30] |
| IGFBP5 | Normal | NA | [30] |
| IGFBP6 | Normal | NA | [30] |
Table 2. Effects on general and kidney phenotypes by gain of function in GH-IGF pathway. There are no data about transgenic models for GHR/GHBP, IGF1R, SOCS, and IGFBP5 and -6.
| General Phenotype | Kidney Phenotype | Ref. | |
|---|---|---|---|
| GH | Giant phenotype, organomegaly | Kidney hypertrophy, glomerular hyperthrophy, progressive albuminuria, glomerulosclerosis | [31][32][33] |
| IGF1 | Enhanced growth | Proportionately enlarged kidneys, glomerular hyperthrophy, no glomerulosclerosis | [34][35][36] |
| IGFBP1 | Low birth weight, postnatal growth retardation, disproportionally small brain, splenomegaly, hyperglycemia | Small kidneys, decreased nephron number; glomerulosclerosis without glomerular hypertrophy | [37][38][39][40] |
| IGFBP2 | Mild growth retardation, mildly reduced organs weight | NA | [41] |
| IGFBP3 | Increased spleen, liver, heart weight | Disproportionally small kidneys | [41][42][43] |
| IGFBP4 | Different tissues hypoplasia | [40] | |
| IGF2 | Disproportionately enlarged kidneys | [44] |
Biallelic mutation in GHRH causes isolated growth hormone deficiency due to impaired GH secretion in anterior hypophysis [2].
GH knockout mice (GH −/−), which show no circulating GH, also show disproportionally reduced kidney weight compared with wild-type mice, even after correction for reduced body weight [19].
GHR/GHPB knockout mice lack functional GH receptors and exhibit GH resistance manifested by decrease in circulating IGF1 levels and growth retardation, starting later after birth. These mice also have disproportionally small kidneys [20].
Germline deletion of Jak2 (downstream of GHR, but also of other hormones and cytokines) in mice resulted in embryonic lethality due to a lack of hematopoiesis [45]. Homozygous mutation in the gene for STAT5 resulted in IGF-1 deficiency and growth hormone insensitivity, indicating impaired postreceptor signaling for GH. It leads to abnormal postnatal growth, facial dysmorphism, and markedly reduced serum concentrations of IGF-1, IGFBP-3, and acid-labile subunit, and immunodeficiency [21]. The latter seems to be due to the importance of both JAK2 and STAT5 not just in mediating GH signals, but also other cytokines involved in immune as well as hematopoetic regulation, such as the erythropoietin receptor [22]. STAT5 knockout mice died perinatally, and 1–2% of survivors were dwarf, with anemia and immunodeficiency [23].
IGF1 knockout mice have severe growth retardation, deficiencies in bone and muscle development, infertility, and lethal respiratory failure due to lung hypoplasia, highlighting the importance of GH/IGF1 axis in different tissues development. Their kidneys are proportionally small with decreased glomerular size and nephron number [25][46].
Prenatal IGF1R knockout embryos exhibit growth retardation and generalized developmental abnormalities, comprising hypoplasia, altered central nervous systems, abnormal skin formation, delayed bone development, reduced pancreatic beta-cells, failure of testicular determination, lung immaturity, and cochlear defects [31]. As IGF1R is closely related to the IR, partly sharing amino acid identity, increased IGF2–mediated IR signaling can rescue mouse embryonic development to prevent dwarfism in IGF1R knockout mice [32].
Mice with homozygous null mutations in Igf1r had normal embryonic development but had low weight and died soon after birth, whereas heterozygous mice had normal growth up to the weaning period, followed by a significant reduction in weight gain and development of insulin resistance [33]. Therefore, the phenotype of the knockout animal is more severe as the location of the affected gene is more distal along the signaling pathway, as described for example for other pathways where the kidneys are a target organ [34].
Excessive GH levels are associated with renal hypertrophy in humans and rodents [35]. Transgenic mice overexpressing the GH gene exhibit excessive GH and IGF1 concentrations, resulting in a giant phenotype and organomegaly, including increased kidney weight even when related to increased body weight [36]. These animals also develop glomerulosclerosis and kidney failure, in association with glomerular hypertrophy and progressive albuminuria [47]. Transgenic mice overexpressing IGF1 are larger than wild-type mice, have proportionately enlarged kidneys [37], and also show glomerular hyperthrophy, but do not develop glomerulosclerosis [38][39]. These findings indicate that GH excess causes glomerular and podocyte hypertrophy sufficient to induce glomerulosclerosis independently of IGF1.
Consistent with the role of IGFBPs as inhibitors of IGF action, their generalized overexpression predominantly results in growth retardation. Mice engineered to overexpress IGFBP-1 have prenatal and postnatal growth retardation, disproportionally small brains, splenomegaly, and glucose intolerance. Their kidneys are proportionally small with a decreased nephron number; they also develop glomerulosclerosis without glomerular hypertrophy [41][42][43][48]. Transgenic mice that overexpress IGFBP-2 have only mild growth retardation, with proportionally small kidneys [30]. Mice overexpressing IGFBP-3 have selective organomegaly (spleen, liver, heart) [28], and disproportionally small kidneys [27], whereas those overexpressing a mutant of IGFBP-3 with impaired IGF binding have normal postnatal growth and kidney size [29], suggesting that the effects on the kidney seen in the former are due to inhibition of IGF actions. IGFBP-4 overexpression in various tissues in mice resulted in hypoplasia of the affected tissue, suggesting a common action in different cell types [40]. Interestingly, only few or no phenotypic changes were observed when separately knocking out each specific IGFBP [49][26][50][51][52].
In the 5/6 nephrectomy mouse model of chronic kidney disease, silencing of SOCS2, a negative regulator of GH action, was shown to overcome CKD-related growth retardation without worsening kidney function. This was explained by elevation of inflammatory cytokines in uremic mice and upregulation of SOCS3, another regulator of cytokine signaling, leading to the prevention of renal GHR overstimulation [53].
5. GH and IGF1 in Normal Renal Development
The GH/IGF1 system plays a key role in normal kidney development, although it does not impair basic kidney formation mediated by the branching morphogenesis process [54]. During embryogenesis, GHR mRNA was detected in rat kidneys from embryonic day 20 and was mainly expressed in the proximal tubules [55]. In the human fetal kidney, GHR-specific immunostaining was shown as early as 8.5 to 9 weeks and most renal tubular epithelial cells became positive by week 13. The staining was stronger in the outer medulla than in the cortex and remained similar at midgestation and after birth. Weak staining was also found in immature glomeruli in early gestation, but disappeared at later developmental stages, suggesting specific GH involvement in glomerular morphogenesis [56].
6. GH/IGF1 Effects on Normal Tubular and Glomerular Functions
Normal kidney function includes glomerular filtration and tubular secretion and reabsorption, leading to fluid and electrolyte balance. In addition, kidneys control blood pressure, as well as hormonal synthesis (such as EPO and active Vitamin D).
GH and IGF1 deficient patients have reduced glomerular filtration rate (GFR) and renal perfusion flow (RPF) [60][61]. Hypophysectomy in humans leads to a rapid decrease in GFR [62], and rhGH treatment leads to GFR and RPF improvement in a dose and time-dependent manner [60][61]. In a cohort of GH-deficient children (isolated or multiple pituitary), GFR was in normal physiological levels but lower than in controls and significantly increased after 3 years of rhGH in parallel to kidney and body growth [63]. In contrast, acromegalic patients have increased GFR and RPF [62][64] and albuminuria [65][66][67] compared with healthy subjects.
GH and IGF1 are involved in tubular handling of sodium, water, calcium, and phosphate, and are also known to regulate tubular gluconeogenesis [68]. GH deficiency is associated with reduced sodium and total body water content [69], and rhGH-replacement therapy improves these parameters [70]. Treatment with high rhGH doses may even lead to acute fluid retention [71]. In contrast to that, acromegalic patients show an increase in total body water and sodium and may present with edema. Treatment of GH-producing tumors reverses these changes [72][73].
The direct, IGF-1-independent effect of GH on sodium and fluid retention is controversial: infused recombinant IGF1 did not change body weight and sodium excretion in healthy subjects [18][74], but treatment with rhIGF1 improved hydration status in children with GH insensitivity due to GHR inactivating mutations, indicating that sodium and water retaining properties of GH are at least partly mediated by IGF1 [75].
Liver-specific deletion of the IGF1 gene increased urinary sodium and potassium excretion [76], confirming the role of IGF1 in water and sodium handling. Evidence for both direct GH/IGF1 action on kidney tubule and indirect mechanisms involving the renin-angiotensin-aldosterone system (RAAS) or natriuretic peptides exists. Rapid increase in plasma renin activity and aldosterone level after rhGH administration in healthy men was reported [77], and treatment with angiotensin converting enzyme (ACE)-inhibitor captopril and mineralocorticoid receptor antagonist spironolactone abolished the GH-induced increase in extracellular volume [78]. Decrease of atrial natriuretic peptide concentration after rhGH treatment was also shown [79]. Recent data show evidence for direct action of GH and IGF1 on epithelial sodium channels (controlled by aldosterone) in cortical collecting ducts [80]. Reversal of GH/IGF1 excess in acromegalic patients decreases ENaC activity [81]. In rats with GH-secreting tumors, the direct stimulatory effect of excess GH on ENaC-dependent sodium transport in distal nephron was demonstrated. Enhanced natriuretic response after ENaC blocking by amiloride and enhanced Na/K-ATPase activity selectively in the cortical collecting ducts were demonstrated, providing additional evidence for increased sodium reabsorption in the late distal nephron during a chronic GH excess. Changes in ENaC subunit proteins, known to be associated with increased ENaC activities [82], were shown in these rats and were not accompanied by elevated aldosterone levels [80]. In humans, active acromegaly was also associated with an increased response to amiloride, providing evidence of increased renal ENaC activity in excess of GH/IGF1 [81]. Another possible molecular target of GH/IGF1 in the kidney tubule is the sodium-potassium pump Na/K-ATPase. GH has been shown to enhance the hydrolytic activity of Na/K-ATPase in rat kidney [83].
Being the major hormones mediating somatic growth, GH and IGF1 promote positive calcium and phosphate balance, influencing, for example, 1.25 (OH)2 vitamin D synthesis, which is crucial for intestinal calcium absorption. GH stimulation of renal calcitriol synthesis is mediated by IGF1 via induction of 1α-hydroxylase in the proximal tubule [84]. GH-replacement therapy, as well as treatment with rhIGF1, increased serum calcitriol levels in GH-deficient patients [85]. Several studies in GH-deficient adults have shown transient elevation in blood calcium level and urinary calcium excretion during rhGH-treatment [86][87]. In contrast, studies in children showed unchanged or even decreased blood calcium levels during long-term rhGH replacement, probably related to modifications of mineral metabolism and a significant increase in bone density [88].
Long-term rhGH treatment leads to a persistent increase in plasma phosphate concentrations in GH-deficient children [85] and adults [83][84][89], which is mediated by a direct antiphosphaturic action of IGF1 in the proximal tubule [90]. IGF1 directly increases phosphate reabsorption via increase of Na-Pi2a expression in proximal tubule, which could be completely blocked by an anti-IGF1R antibody [91][92]. Patients with acromegaly may have mild hyperphosphatemia that normalizes after treatment of their GH-secreting tumor [93].
The physiologic roles of GH and IGF1 in different nephron segments are depicted in Figure 1.
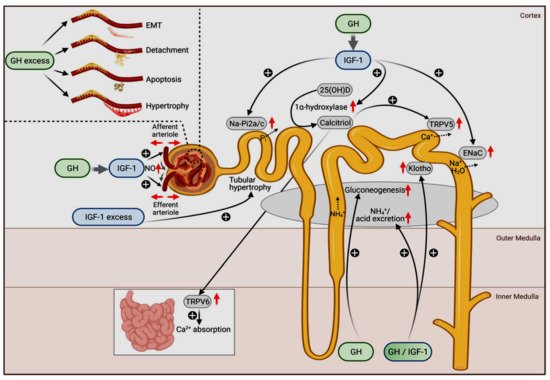
Figure 1. Physiological (main figure) and pathophysiological actions of GH (upper left insert) and IGF-1 on the kidneys. The original figure has been published by Hafner et al. [94] and published here with permission. The figure is licensed under a Creative Commons Attribution 4.0 International License. See link to the Creative Commons license (http://creativecommons.org/licenses/by/4.0/, accessed on 29 November 2021). No changes to the original figure were made.
References
- Hartman, M.L.; Veldhuis, J.D.; Thorner, M.O. Normal control of growth hormone secretion. Horm. Res. 1993, 40, 37–47.
- Pang, A.L.-Y.; Chan, W.Y. Chapter 22—Molecular Basis of Diseases of the Endocrine System. In Molecular Pathology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2018.
- Rubinek, T.; Modan-Moses, D. Klotho and the growth hormone/insulin-like growth factor 1 axis: Novel insights into complex interactions. Vitam. Horm. 2016, 101, 85–118.
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998, 93, 385–395.
- Hansen, J.A.; Lindberg, K.; Hilton, D.J.; Nielsen, J.H.; Billestrup, N. Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol. Endocrinol. 1999, 13, 1832–1843.
- Frystyk, J.; Skjaerbaek, C.; Dinesen, B.; Orskov, H. Free insulin-like growth factors (IGF-I and IGF-II) in human serum. FEBS Lett. 1994, 348, 185–191.
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96.
- Werner, H.; Bruchim, I. The insulin-like growth factor-I receptor as an oncogene. Arch. Physiol. Biochem. 2009, 115, 58–71.
- Baxter, R.C. Insulin-like growth factor (IGF)-binding proteins: Interactions with IGFs and intrinsic bioactivities. Am. J. Physiol. Metab. 2000, 278, E967–E976.
- Mohan, S.; Baylink, D.J. IGF-binding proteins are multifunctional and act via IGF-dependent and -independent mechanisms. J. Endocrinol. 2002, 175, 19–31.
- Salmon, W.D., Jr.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836.
- Green, H.; Morikawa, M.; Nixon, T. A dual effector theory of growth-hormone action. Differentiation 1985, 29, 195–198.
- Yakar, S.; Liu, J.L.; Le Roith, D. The growth hormone/insulin-like growth factor-I system: Implications for organ growth and development. Pediatr. Nephrol. 2000, 14, 544–549.
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The somatomedin hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74.
- Roberts, C.T., Jr.; Lasky, S.R.; Lowe, W.L., Jr.; Seaman, W.T.; LeRoith, D. Molecular cloning of rat insulin-like growth factor I complementary deoxyribonucleic acids: Differential messenger ribonucleic acid processing and regulation by growth hormone in extrahepatic tissues. Mol. Endocrinol. 1987, 1, 243–248.
- Schimpff, R.M.; Donnadieu, M.; Duval, M. Serum somatomedin activity measured as sulphation factor in peripheral, hepatic and renal veins in normal mongrel dogs: Early effects of intravenous injection of growth hormone. Acta Endocrinol. 1980, 93, 155–161.
- Guler, H.P.; Zapf, J.; Scheiwiller, E.; Froesch, E.R. Recombinant human insulin-like growth factor I stimulates growth and has distinct effects on organ size in hypophysectomized rats. Proc. Natl. Acad. Sci. USA 1988, 85, 4889–4893.
- Guler, H.P.; Schmid, C.; Zapf, J.; Froesch, E.R. Effects of recombinant insulin-like growth factor I on insulin secretion and renal function in normal human subjects. Proc. Natl. Acad. Sci. USA 1989, 86, 2868–2872.
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162.
- List, E.O.; Sackmann-Sala, L.; Berryman, D.E.; Funk, K.; Kelder, B.; Gosney, E.S.; Okada, S.; Ding, J.; Cruz-Topete, D.; Kopchick, J.J. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR−/−) mouse. Endocr. Rev. 2011, 32, 356–386.
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Jasper, H.; Tepper, A.; et al. Growth hormone insensitivity associated with a STAT5b mutation. N. Engl. J. Med. 2003, 349, 1139–1147.
- Landau, D.; London, L.; Bandach, I.; Segev, Y. The hypoxia inducible factor/erythropoietin (EPO)/EPO receptor pathway is disturbed in a rat model of chronic kidney disease related anemia. PLoS ONE 2018, 13, e0196684.
- Cui, Y.; Riedlinger, G.; Miyoshi, K.; Tang, W.; Li, C.; Deng, C.X.; Robinson, G.W.; Hennighausen, L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol. Cell. Biol. 2004, 24, 8037–8047.
- Mulroney, S.E.; Woda, C.; Johnson, M.; Pesce, C. Gender differences in renal growth and function after uninephrectomy in adult rats. Kidney Int. 1999, 56, 944–953.
- Liu, J.L.; Yakar, S.; LeRoith, D. Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system. Proc. Soc. Exp. Boil. Med. 2000, 223, 344–351.
- Wood, T.L.; Rogler, L.E.; Czick, M.E.; Schuller, A.G.; Pintar, J.E. Selective alterations in organ sizes in mice with a targeted disruption of the insulin-like growth factor binding protein-2 gene. Mol. Endocrinol. 2000, 14, 1472–1482.
- Modric, T.; Silha, J.V.; Shi, Z.; Gui, Y.; Suwanichkul, A.; Durham, S.K.; Powell, D.R.; Murphy, L.J. Phenotypic manifestations of insulin-like growth factor-binding protein-3 overexpression in transgenic mice. Endocrinology 2001, 142, 1958–1967.
- Murphy, L.J.; Molnar, P.; Lu, X.; Huang, H. Expression of human insulin-like growth factor-binding protein-3 in transgenic mice. J. Mol. Endocrinol. 1995, 15, 293–303.
- Silha, J.V.; Gui, Y.; Mishra, S.; Leckstrom, A.; Cohen, P.; Murphy, L.J. Overexpression of gly56/gly80/gly81-mutant insulin-like growth factor-binding protein-3 in transgenic mice. Endocrinology 2005, 146, 1523–1531.
- Hoeflich, A.; Wu, M.; Mohan, S.; Föll, J.; Wanke, R.; Froehlich, T.; Arnold, G.J.; Lahm, H.; Kolb, H.J.; Wolf, E. Overexpression of insulin-like growth factor-binding protein-2 in transgenic mice reduces postnatal body weight gain. Endocrinology 1999, 140, 5488–5496.
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72.
- Louvi, A.; Accili, D.; Efstratiadis, A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev. Biol. 1997, 189, 33–48.
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187.
- Rotem-Grunbaum, B.; Landau, D. Genetic renal disease classification by hormonal axes. Pediatr. Nephrol. 2020, 35, 2211–2219.
- Kamenický, P.; Mazziotti, G.; Lombès, M.; Giustina, A.; Chanson, P. Growth hormone, insulin-like growth factor-1, and the kidney: Pathophysiological and clinical implications. Endocr. Rev. 2014, 35, 234–281.
- Striker, L.J.; Doi, T.; Striker, G.E. Transgenic mice in renal research. Adv. Nephrol. Necker Hosp. 1991, 20, 91–108.
- Mathews, L.S.; Hammer, R.E.; Behringer, R.R.; D’Ercole, A.J.; Bell, G.I.; Brinster, R.L.; Palmiter, R.D. Growth enhancement of transgenic mice expressing human insulin-like growth factor I. Endocrinology 1988, 123, 2827–2833.
- Doi, T.; Striker, L.J.; Quaife, C.; Conti, F.G.; Palmiter, R.; Behringer, R.; Brinster, R.; Striker, G.E. Progressive glomerulosclerosis develops in transgenic mice chronically expressing growth hormone and growth hormone releasing factor but not in those expressing insulin like growth factor-1. Am. J. Pathol. 1988, 131, 398–403.
- Doi, T.; Striker, L.J.; Gibson, C.C.; Agodoa, L.Y.; Brinster, R.L.; Striker, G.E. Glomerular lesions in mice transgenic for growth hormone and insulin like growth factor-I. I. Relationship between increased glomerular size and mesangial sclerosis. Am. J. Pathol. 1990, 137, 541–552.
- Blutke, A.; Schneider, M.R.; Wolf, E.; Wanke, R. Growth hormone (GH)-transgenic insulin-like growth factor 1 (IGF1)-deficient mice allow dissociation of excess GH and IGF1 effects on glomerular and tubular growth. Physiol. Rep. 2016, 4, e12709.
- Rajkumar, K.; Barron, D.; Lewitt, M.S.; Murphy, L.J. Growth retardation and hyperglycemia in insulin-like growth factor binding protein-1 transgenic mice. Endocrinology 1995, 136, 4029–4034.
- Doublier, S.; Seurin, D.; Fouqueray, B.; Verpont, M.C.; Callard, P.; Striker, L.J.; Striker, G.E.; Binoux, M.; Baud, L. Glomerulosclerosis in mice transgenic for human insulin-like growth factor-binding protein-1. Kidney Int. 2000, 57, 2299–2307.
- Doublier, S.; Amri, K.; Seurin, D.; Moreau, E.; Merlet-Benichou, C.; Striker, G.E.; Gilbert, T. Overexpression of human insulin-like growth factor binding protein-1 in the mouse leads to nephron deficit. Pediatr. Res. 2001, 49, 660–666.
- Lindenbergh-Kortleve, D.J.; Rosato, R.R.; van Neck, J.W.; Nauta, J.; van Kleffens, M.; Groffen, C.; Zwarthoff, E.C.; Drop, S.L. Gene expression of the insulin-like growth factor system during mouse kidney development. Mol. Cell. Endocrinol. 1997, 132, 81–91.
- Neubauer, H.; Cumano, A.; Müller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998, 93, 397–409.
- Rogers, S.A.; Powell-Braxton, L.; Hammerman, M.R. Insulin-like growth factor I regulates renal development in rodents. Dev. Genet. 1999, 24, 293–298.
- Pesce, C.M.; Striker, L.J.; Peten, E.; Elliot, S.J.; Striker, G.E. Glomerulosclerosis at both early and late stages is associated with increased cell turnover in mice transgenic for growth hormone. Lab. Investig. 1991, 65, 601–605.
- Schneider, M.R.; Lahm, H.; Wu, M.; Hoeflich, A.; Wolf, E. Transgenic mouse models for studying the functions of insulin-like growth factor-binding proteins. FASEB J. 2000, 14, 629–640.
- Ning, Y.; Schuller, A.G.; Bradshaw, S.; Rotwein, P.; Ludwig, T.; Frystyk, J.; Pintar, J.E. Diminished growth and enhanced glucose metabolism in triple knockout mice containing mutations of insulin-like growth factor binding protein-3, -4, and -5. Mol. Endocrinol. 2006, 20, 2173–2186.
- Leu, J.I.; Crissey, M.A.; Craig, L.E.; Taub, R. Impaired hepatocyte DNA synthetic response posthepatectomy in insulin-like growth factor binding protein 1-deficient mice with defects in C/EBP beta and mitogen-activated protein kinase/extracellular signal-regulated kinase regulation. Mol. Cell. Biol. 2003, 23, 1251–1259.
- DeMambro, V.E.; Clemmons, D.R.; Horton, L.G.; Bouxsein, M.L.; Wood, T.L.; Beamer, W.G.; Canalis, E.; Rosen, C.J. Gender-specific changes in bone turnover and skeletal architecture in igfbp-2-null mice. Endocrinology 2008, 149, 2051–2061.
- Gray, A.; Aronson, W.J.; Barnard, R.J.; Mehta, H.; Wan, J.; Said, J.; Cohen, P.; Galet, C. Global Igfbp1 deletion does not affect prostate cancer development in a c-Myc transgenic mouse model. J. Endocrinol. 2011, 211, 297–304.
- Landau, D.; Assadi, M.H.; Abu Hilal, R.; Chen, Y.; Rabkin, R.; Segev, Y. SOCS2 Silencing Improves Somatic Growth without Worsening Kidney Function in CKD. Am. J. Nephrol. 2020, 51, 520–526.
- Lindström, N.O.; McMahon, J.A.; Guo, J.; Tran, T.; Guo, Q.; Rutledge, E.; Parvez, R.K.; Saribekyan, G.; Schuler, R.E.; Liao, C.; et al. Conserved and divergent features of human and mouse kidney organogenesis. J. Am. Soc. Nephrol. 2018, 29, 785–805.
- Chin, E.; Zhou, J.; Bondy, C.A. Renal growth hormone receptor gene expression: Relationship to renal insulin-like growth factor system. Endocrinology 1992, 131, 3061–3066.
- Simard, M.; Manthos, H.; Giaid, A.; Lefèbvre, Y.; Goodyer, C.G. Ontogeny of growth hormone receptors in human tissues: An immunohistochemical study. J. Clin. Endocrinol. Metab. 1996, 81, 3097–3102.
- Rogers, S.A.; Ryan, G.; Hammerman, M.R. Insulin-like growth factors I and II are produced in the metanephros and are required for growth and development in vitro. J. Cell Biol. 1991, 113, 1447–1453.
- Daughaday, W.H.; Rotwein, P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 1989, 10, 68–91.
- Wolf, E.; Kramer, R.; Blum, W.F.; Föll, J.; Brem, G. Consequences of postnatally elevated insulin-like growth factor-II in transgenic mice: Endocrine changes and effects on body and organ growth. Endocrinology 1994, 135, 1877–1886.
- Caidahl, K.; Edén, S.; Bengtsson, B.A. Cardiovascular and renal effects of growth hormone. Clin. Endocrinol. 1994, 40, 393–400.
- Jørgensen, J.O.; Pedersen, S.A.; Thuesen, L.; Jørgensen, J.; Ingemann-Hansen, T.; Skakkebaek, N.E.; Christiansen, J.S. Beneficial effects of growth hormone treatment in GH-deficient adults. Lancet 1989, 333, 1221–1225.
- Falkheden, T.; Sjoegren, B. Extracellular fluid volume and renal function in pituitary insufficiency and acromegaly. Acta Endocrinol. 1964, 46, 80–88.
- Ece, A.; Çetinkaya, S.; Ekşioğlu, S.; Şenel, S.; Özkasap, S.; Giniş, T.; Sen, V.; Şahin, C. Kidney growth and renal functions under the growth hormone replacement therapy in children. Ren. Fail. 2014, 36, 508–513.
- Ikkos, D.; Ljunggren, H.; Luft, R. Glomerular filtration rate and renal plasma flow in acromegaly. Acta Endocrinol. 1956, 21, 226–236.
- Grunenwald, S.; Tack, I.; Chauveau, D.; Bennet, A.; Caron, P. Impact of growth hormone hypersecretion on the adult human kidney. Ann. Endocrinol. 2011, 72, 485–495.
- Hoogenberg, K.; Sluiter, W.J.; Dullaart, R.P. Effect of growth hormone and insulin-like growth factor I on urinary albumin excretion: Studies in acromegaly and growth hormone deficiency. Acta Endocrinol. 1993, 129, 151–157.
- Manelli, F.; Bossoni, S.; Burattin, A.; Doga, M.; Solerte, S.B.; Romanelli, G.; Giustina, A. Exercise-induced microalbuminuria in patients with active acromegaly: Acute effects of slow-release lanreotide, a long-acting somatostatin analog. Metabolism 2000, 49, 634–639.
- Feld, S.; Hirschberg, R. Growth hormone, the insulin-like growth factor system, and the kidney. Endocr. Rev. 1996, 17, 423–480.
- De Boer, H.; Blok, G.J.; Van der Veen, E.A. Clinical aspects of growth hormone deficiency in adults. Endocr. Rev. 1995, 16, 63–86.
- Jørgensen, J.O. Human growth hormone replacement therapy: Pharmacological and clinical aspects. Endocr. Rev. 1991, 12, 189–207.
- Boguszewski, M.C.S. Growth hormone deficiency and replacement in children. Rev. Endocr. Metab. Disord. 2021, 22, 101–108.
- Ikkos, D.; Luft, R.; Sjogren, B. Body water and sodium in patients with acromegaly. J. Clin. Investig. 1954, 33, 989–994.
- Kamenický, P.; Maione, L.; Chanson, P. Cardiovascular complications of acromegaly. Ann. Endocrinol. 2020, 82, 206–209.
- Hirschberg, R.; Brunori, G.; Kopple, J.D.; Guler, H.P. Effects of insulin-like growth factor I on renal function in normal men. Kidney Int. 1993, 43, 387–397.
- Walker, J.L.; Ginalska-Malinowska, M.; Romer, T.E.; Pucilowska, J.B.; Underwood, L.E. Effects of the infusion of insulin-like growth factor I in a child with growth hormone insensitivity syndrome (Laron dwarfism). N. Engl. J. Med. 1991, 324, 1483–1488.
- Svensson, J.; Tivesten, A.; Sjögren, K.; Isaksson, O.; Bergström, G.; Mohan, S.; Mölne, J.; Isgaard, J.; Ohlsson, C. Liver-derived IGF-I regulates kidney size, sodium reabsorption, and renal IGF-II expression. J. Endocrinol. 2007, 193, 359–366.
- Ho, K.Y.; Weissberger, A.J. The antinatriuretic action of biosynthetic human growth hormone in man involves activation of the renin-angiotensin system. Metabolism 1990, 39, 133–137.
- Møller, J.; Møller, N.; Frandsen, E.; Wolthers, T.; Jørgensen, J.O.; Christiansen, J.S. Blockade of the renin-angiotensin-aldosterone system prevents growth hormone-induced fluid retention in humans. Am. J. Physiol. Content 1997, 272, E803–E808.
- Møller, J.; Jørgensen, J.O.; Møller, N.; Hansen, K.W.; Pedersen, E.B.; Christiansen, J.S. Expansion of extracellular volume and suppression of atrial natriuretic peptide after growth hormone administration in normal man. J. Clin. Endocrinol. Metab. 1991, 72, 768–772.
- Kamenicky, P.; Viengchareun, S.; Blanchard, A.; Meduri, G.; Zizzari, P.; Imbert-Teboul, M.; Doucet, A.; Chanson, P.; Lombès, M. Epithelial sodium channel is a key mediator of growth hormone-induced sodium retention in acromegaly. Endocrinology 2008, 149, 3294–3305.
- Kamenicky, P.; Blanchard, A.; Frank, M.; Salenave, S.; Letierce, A.; Azizi, M.; Lombès, M.; Chanson, P. Body fluid expansion in acromegaly is related to enhanced epithelial sodium channel (ENaC) activity. J. Clin. Endocrinol. Metab. 2011, 96, 2127–2135.
- Hughey, R.P.; Bruns, J.B.; Kinlough, C.L.; Harkleroad, K.L.; Tong, Q.; Carattino, M.D.; Johnson, J.P.; Stockand, J.D.; Kleyman, T.R. Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 2004, 279, 18111–18114.
- Shimomura, Y.; Lee, M.; Oku, J.; Bray, G.A.; Glick, Z. Sodium potassium dependent ATPase in hypophysectomized rats: Response to growth hormone, triiodothyronine, and cortisone. Metabolism 1982, 31, 213–216.
- Nesbitt, T.; Drezner, M.K. Insulin-like growth factor-I regulation of renal 25-hydroxyvitamin D-1-hydroxylase activity. Endocrinology 1993, 132, 133–138.
- Bianda, T.; Glatz, Y.; Bouillon, R.; Froesch, E.R.; Schmid, C. Effects of short-term insulin-like growth factor-I (IGF-I) or growth hormone (GH) treatment on bone metabolism and on production of 1,25-dihydroxycholecalciferol in GH-deficient adults. J. Clin. Endocrinol. Metab. 1998, 83, 81–87.
- Bengtsson, B.A.; Edén, S.; Lönn, L.; Kvist, H.; Stokland, A.; Lindstedt, G.; Bosaeus, I.; Tölli, J.; Sjöström, L.; Isaksson, O.G. Treatment of adults with growth hormone (GH) deficiency with recombinant human GH. J. Clin. Endocrinol. Metab. 1993, 76, 309–317.
- Hansen, T.B.; Brixen, K.; Vahl, N.; Jørgensen, J.O.; Christiansen, J.S.; Mosekilde, L.; Hagen, C. Effects of 12 months of growth hormone (GH) treatment on calciotropic hormones, calcium homeostasis, and bone metabolism in adults with acquired GH deficiency: A double blind, randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 1996, 81, 3352–3359.
- Saggese, G.; Baroncelli, G.I.; Bertelloni, S.; Cinquanta, L.; Di Nero, G. Effects of long-term treatment with growth hormone on bone and mineral metabolism in children with growth hormone deficiency. J. Pediatr. 1993, 122, 37–45.
- Ahmad, A.M.; Thomas, J.; Clewes, A.; Hopkins, M.T.; Guzder, R.; Ibrahim, H.; Durham, B.H.; Vora, J.P.; Fraser, W.D. Effects of growth hormone replacement on parathyroid hormone sensitivity and bone mineral metabolism. J. Clin. Endocrinol. Metab. 2003, 88, 2860–2868.
- Quigley, R.; Baum, M. Effects of growth hormone and insulin-like growth factor I on rabbit proximal convoluted tubule transport. J. Clin. Investig. 1991, 88, 368–374.
- Hirschberg, R.; Ding, H.; Wanner, C. Effects of insulin-like growth factor I on phosphate transport in cultured proximal tubule cells. J. Lab. Clin. Med. 1995, 126, 428–434.
- Jehle, A.W.; Forgo, J.; Biber, J.; Lederer, E.; Krapf, R.; Murer, H. IGF-I and vanadate stimulate Na/Pi-cotransport in OK cells by increasing type II Na/Pi-cotransporter protein stability. Pflugers Arch. 1998, 437, 149–154.
- Xie, T.; Tian, P.; Wu, S.; Zhang, X.; Liu, T.; Gu, Y.; Sun, C.; Hu, F. Serum phosphate: Does it more closely reflect the true state of acromegaly? J. Clin. Neurosci. 2020, 71, 26–31.
- Haffner, D.; Grund, A.; Leifheit-Nestler, M. Renal effects of growth hormone in health and in kidney disease. Pediatr. Nephrol. 2021, 36, 2511–2530.
More
Information
Subjects:
Psychology, Biological
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
4 times
(View History)
Update Date:
17 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No