 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chaofan Fan | + 2110 word(s) | 2110 | 2021-11-03 06:59:42 |
Video Upload Options
Metabolic reprogramming and epigenetic changes have been characterized as hallmarks of liver cancer. Metabolic intermediates serve as crucial substrates for various epigenetic modulations, from post-translational modification of histones to DNA methylation. In turn, epigenetic changes can alter the expression of metabolic genes supporting on the one hand, the increased energetic demand of cancer cells and, on the other hand, influence the activity of tumor-associated immune cell populations. In this review, we will illustrate the most recent findings about metabolic reprogramming in liver cancer. We will focus on the metabolic changes characterizing the tumor microenvironment and on how these alterations impact on epigenetic mechanisms involved in the malignant progression. Furthermore, we will report our current knowledge about the influence of cancer-specific metabolites on epigenetic reprogramming of immune cells. Finally, we will review the current strategies to target metabolic and epigenetic pathways and their therapeutic potential in liver cancer, alone or in combinatorial approaches.
1. Introduction
Liver cancer is the fourth most common cause of cancer death worldwide [1]. No effective liver cancer therapy has been approved so far and therefore there is an urgent need to find efficient and safe therapeutic strategies. Liver cancer is induced by several pathological processes normally related to chronic inflammation, such as metabolic and nutritional disturbances (ASH/NASH) as well as viral infections (HBV/HCV) [2]. Generally, conditions leading to hepatocyte stress or damage inevitably result in alterations of their metabolic activity. Vice-versa, loss of the energetic balance could impair the functionality of cellular organelles leading to hepatocyte damage. Accordingly, the oncogenic transformation of hepatocytes towards a malignant phenotype is characterized by dramatic changes of the cellular metabolism referred to as metabolic reprogramming. It is nowadays clear from experimental models and clinical evidence that many oncogenic mutations selectively induce metabolic changes that critically sustain tumor growth. However, in liver cancer, not only the genomic landscape but also the immune characteristics of transforming cells frequently define the microenvironment that dictates a distinct metabolic profile of the tumor, as, for instance, in the case of NASH-related HCC [3]. In turn, metabolites produced by cancer cells generate a permissive immune-microenvironment for favorable growing conditions. Alterations of the cellular metabolism occurring in transforming malignant cells markedly influence the epigenetic mechanisms and, in return, epigenetic modifications commonly sustain the metabolic reprogramming through the regulation of specific genes [4].
In this review, we describe the most important changes of the metabolism characterizing and supporting hepatocarcinogenesis. We will illustrate how these changes of cellular metabolism impact on epigenetic modifications highlighting the most recent findings about the major alterations of the chromatin occurring in liver cancer cells. Successively, we will describe how metabolic reprogramming of liver cancer can possibly influence the tumor immune-microenvironment and how epigenetic alterations of the immune cells can affect cancer growth. Finally, we will review the most novel findings about current therapeutic strategies aiming to modulate cancer-specific metabolic pathways and epigenetic modifications in order to render cancer more vulnerable to conventional therapies.
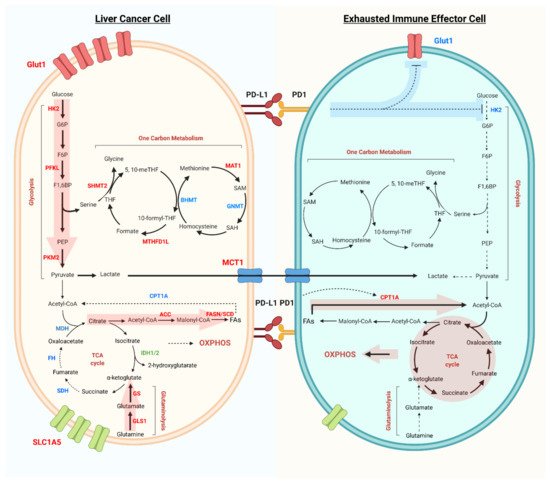
2. Metabolic Reprogramming in Liver Cancer
2.1. Glycolysis
2.2. TCA Cycle and Lipid Metabolism
2.3. Amino Acids Metabolism
2.4. One-Carbon Metabolism
3. Metabolic Reprogramming Leading to Epigenetic Changes in Tumor Cells
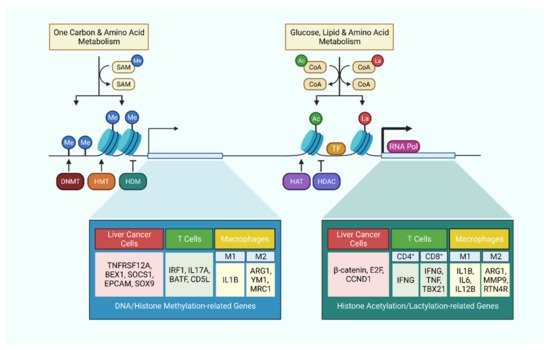
3.1. DNA Methylation
3.2. Histone Modifications
3.3. ATP-Dependent Chromatin Remodeling
4. Metabolism of the Immune Cells in the Microenvironment of Liver Cancer
The discussed tumor metabolic reprogramming gives rise to a suboptimal metabolic microenvironment for infiltrated immune cells. These conditions have a pivotal role in driving the exhaustion of immune effector cells, in particular CD8+ T cell and NK cell exhaustion, which in turn provokes immune evasion (Figure 1).
4.1. Glycolysis
T cell receptor (TCR) dependent CD8+ T cells activation involves an initial switch from oxidative phosphorylation to glycolysis [24]. This short-term metabolic reprogramming boosts the early translation of IFN-γ, TNF-α, and IL-2 transcripts. Thereafter, long-term glycolytic reprogramming is induced by co-stimulatory signaling of CD28, via the Akt/mTOR and HIF-1α pathway to promote Glut1-dependent glucose uptake as well as the expression of glycolytic enzymes, which sustains the increasing energetic and biosynthetic demands. Due to the biological importance of the glycolytic pathway, scarcity of glucose in the TME imposes a major hurdle for effector T cell activity. Sole restriction of glucose availability limits CD8+ T cell responsiveness, despite the presence of tumor antigenic stimulation [25].
4.2. TCA Cycle, Glutaminolysis, and Fatty Acid Oxidation
As discussed, deprivation of glucose in the TME and immune checkpoint signaling can both suppress glycolysis, forcing immune cells to switch to fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS). The overexpression of the FAO rate-limiting enzyme CPT1A by PD-1 signaling has been shown to be responsible for such a shift in energy production. Loss of mitochondrial function results in the subsequent elevation of intra-cellular ROS level, which prevents the dephosphorylation of NFAT, causing the persistent signaling that eventually drives T cell exhaustion [26]. Conversely, regulatory T cells (Treg) appear to be able to thrive in the TME, despite of the adverse nutritional context.
Glutamine is one of the major carbon sources in cells, and it fuels the TCA cycle in the form of α-KG generated from glutaminolysis. Abolishment of this catabolic process skews macrophage polarization towards the M1 phenotype, and leads to the reduction of myeloid-derived suppressor cells (MDSCs), rendering the tumor more susceptible to immune checkpoint therapy [27].
4.3. One-Carbon Metabolism
Methionine influx and metabolism are crucial for T cell activation upon stimulation of antigen receptors, nucleic acids, and reducing agents, together with crucial precursors and co-factors to maintain critical biological processes [28]. GSH is required to mitigate the ROS triggered by TCR-mediated T cell activation. This antioxidant mechanism upregulates MYC production via the NFAT-mTOR axis to initiate reprogramming of glucose metabolism and facilitate T cell proliferation and cytokine secretion [29].
5. Metabolism-Regulated Immunoepigenetic Changes in Liver Cancer
Similar to the case of cancer cells, the aforementioned metabolic aberrations also contribute to the epigenetic reprogramming of immune cells. Here, we highlight the epigenetic changes of immune cells under the context of tumor-induced metabolic reprogramming.
5.1. Glycolysis and TCA Cycle-Associated Epigenetic Modifications
Acetyl-CoA is a metabolic intermediate connecting glycolysis to the TCA cycle. TLR activation in macrophages induces upregulation of glycolysis and ATP citrate lyase activity to yield acetyl-CoA, which facilitates the histone H3 and H4 acetylation of specific innate immune responsive gene sets to increase enhancer accessibility [30]. A similar mechanism was also observed in CD4+ T cells, where activated T cells upregulate LDHA expression to maximize aerobic glycolysis. This results in histone H3 acetylation of both the promoter and enhancer of IFN-γ, and ultimately promotes effector T cell differentiation [106]. Under glucose deprivation, CD8+ T cells were shown to uptake extracellular acetate through MCT-1 and MCT4, and then converted it into acetyl-CoA by ACSS2 for histone H3 and H4 acetylation of cytokine genes to maintain effector function in adverse conditions [31].
5.2. One-Carbon Metabolism-Associated Epigenetic Modifications
The metabolite intermediate of the methionine cycle, SAM, plays a major role as the universal methyl donor in DNA and histone methylation. For instance, in HCC, high concentration of SAM and MTA leads to lowered accessibility of key T cell activation gene sets which are involved in lymphocyte proliferation and differentiation, and hence skewing towards an exhaustion phenotype [32].
6. Metabolic Targets in the Treatment of Liver Cancer
In the last decade, many studies have been focused on the development of metabolic inhibitors as supplemental approach for cancer therapy. Here, we discuss the recent advancement of metabolic strategies for liver cancer and anti-tumor immunity (Table 2).
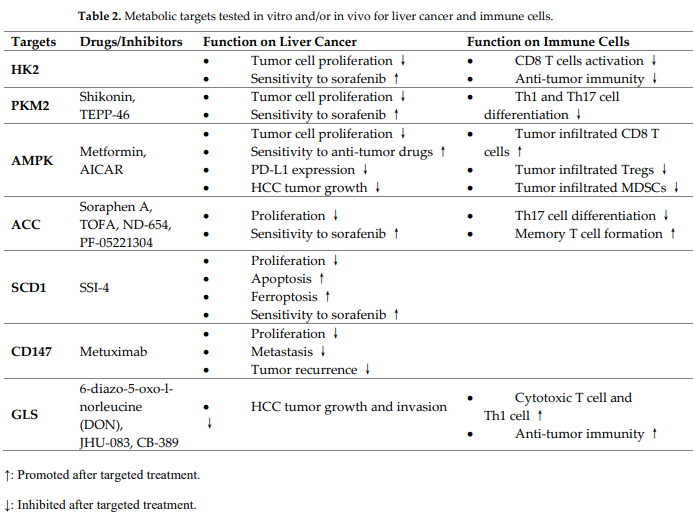
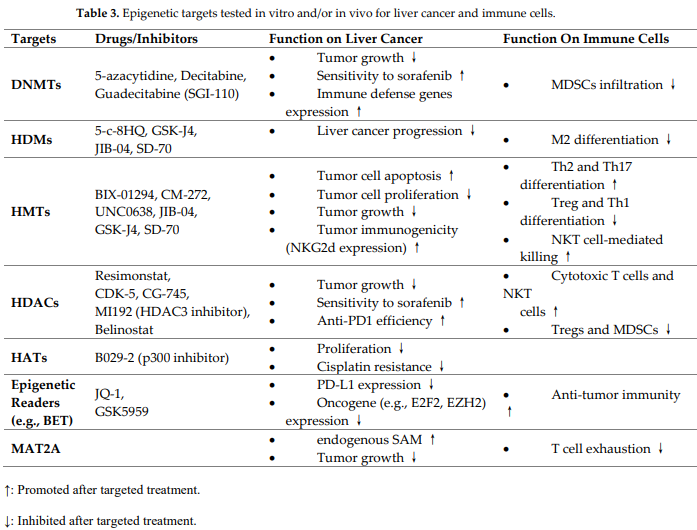
7. Epigenetic Targets in the Treatment of Liver Cancer
In the liver, mutations and altered expression of epigenetic modifiers in relation to driven metabolic rewiring are common, and tackling these epigenetic changes can be exploited to increase the efficacy of therapies against liver cancer [33]. Current epigenetic-regulating strategies for liver cancer therapy are reviewed here (Table 3).
8. Conclusions and Outlook
To date, liver cancer remains as one of the deadliest cancer types, and its global incidence rate continues to grow steadily. The current therapeutic options for advanced liver cancer are limited and often shows unsatisfactory benefits in patients. With the development and testing of novel metabolic and epigenetic inhibitors, it opens up new opportunities to interfere with cancer metabolism, epigenetic dysregulation, and normalize the immune microenvironment to enhance potentially anti-cancer immunity.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F.; Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. . CA Cancer J. Clin. 2021, 71, 209-249, https://doi.org/10.3322/caac.21660..
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S.; et al. Hepatocellular carcinoma.. Nat. Rev. Dis. Primers 2021, 7, 6.
- Pinyol, R., Torrecilla, S.; Wang, H.; Montironi, C.; Pique-Gili, M.; Torres-Martin, M.; Wei-Qiang, L.; Willoughby, C.E.; Ramadori, P.; Andreu-Oller, C.; et al.; et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis.. J. Hepatol. 2021, 75, 865-878.
- Xiong, L.; Wu, F.; Wu, Q.; Xu, L.; Cheung, O.K.; Kang, W.; Mok, M.T.; Szeto, L.L.M.; Lun, C.-Y.; Lung, R.W.; et al.et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional repro-gramming. . Nat. Commun. 2019, 10, 335, https://doi.org/10.1038/s41467-018-08245-z..
- Yang, Y.; Zhang, G.; Guo, F.; Li, Q.; Luo, H.; Shu, Y.; Shen, Y.; Gan, J.; Xu, L.; Yang, H.; et al. Mitochondrial UQCC3 Modulates Hypoxia Adaptation by Orchestrating OXPHOS and Glycolysis in Hepatocellu-lar Carcinoma. . Cell Rep. 2020, 33, 108340, https://doi.org/10.1016/j.celrep.2020.108340..
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C.; High Expression of Glycolytic Genes in Cirrhosis Correlates With the Risk of Developing Liver Cancer. . Front. Cell Dev. Biol. 2018, 6, 138, https://doi.org/10.3389/fcell.2018.00138..
- Shang, R.; Pu, M.; Li, Y.; Wang, D.; FOXM1 regulates glycolysis in hepatocellular carcinoma by transactivating glucose transporter 1 expression.. Oncol. Rep. 2017, 37, 2261-2269, https://doi.org/10.3892/or.2017.5472..
- Lulli, M.; Del Coco, L.; Mello, T.; Sukowati, C.; Madiai, S.; Gragnani, L.; Forte, P.; Fanizzi, F.P.; Mazzocca, A.; Rom-bouts, K.; et al.et al. DNA Damage Response Protein CHK2 Regulates Metabolism in Liver Cancer.. Cancer Res. 2021, 81, 2861-2873, https://doi.org/10.1158/0008-5472.can-20-3134..
- Lee, K.; Song, Y.S.; Shin, Y.; Wen, X.; Kim, Y.; Cho, N.Y.; Bae, J.M.; Kang, G.H.; Intrahepatic cholangiocarcinomas with IDH1/2 mutation-associated hypermethylation at selective genes and their clinicopathological features. . Sci. Rep. 2020, 10, 15820.
- Budhu, A.; Roessler, S.; Zhao, X.; Yu, Z.; Forgues, M.; Ji, J.; Karoly, E.; Qin, L.; Ye, Q.; Jia, H.; et al.et al. Integrated Metabolite and Gene Expression Profiles Identify Lipid Biomarkers Associated With Progression of Hepatocellular Carcinoma and Patient Outcomes.. Gastroenterology 2013, 144, 1066-1075.e1, https://doi.org/10.1053/j.gastro.2013.01.054..
- Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Simile, M.M.; Ribback, S.; Dombrowski, F.; Evert, M.; Chen, X.; Calvisi, D.F.; et al. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. . Cell Cycle 2017, 16, 499-507, https://doi.org/10.1080/15384101.2017.1282586..
- Senni, N.; Savall, M.; Granados, D.C.; Alves-Guerra, M.C.; Sartor, C.; Lagoutte, I.; Gougelet, A.; Terris, B.; Gilgen-krantz, H.; Perret, C.; et al.et al. Beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. . Gut 2019, 68, 322-334.
- Nilsson, A.; Haanstra, J.R.; Engqvist, M.; Gerding, A.; Bakker, B.M.; Klingmüller, U.; Teusink, B.; Nielsen, J.; Quantitative analysis of amino acid metabolism in liver cancer links glutamate excretion to nucleotide synthesis. . Proc. Natl. Acad. Sci. USA 2020, 117, 10294-10304, https://doi.org/10.1073/pnas.1919250117..
- Tompkins, S.C.; Sheldon, R.; Rauckhorst, A.J.; Noterman, M.; Solst, S.R.; Buchanan, J.L.; Mapuskar, K.A.; Pewa, A.D.; Gray, L.R.; Oonthonpan, L.; et al.et al. Disrupting Mitochondrial Pyruvate Uptake Directs Glutamine into the TCA Cycle away from Glutathione Synthe-sis and Impairs Hepatocellular Tumorigenesis.. Cell Rep. 2019, 28, 2608-2619, https://doi.org/10.1016/j.celrep.2019.07.098..
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.; Couchy, G.; Laurent, A.; Ba-labaud, C.; et al.et al. Genotype‐phenotype correlation of CTNNB1 mutations reveals different ß‐catenin activity associated with liver tumor progression.. Hepatology 2016, 64, 2047-2061, https://doi.org/10.1002/hep.28638..
- Anderton, B.; Camarda, R.; Balakrishnan, S.; Balakrishnan, A.; A Kohnz, R.; Lim, L.; Evason, K.J.; Momcilovic, O.; Kruttwig, K.; Huang, Q.; et al.et al. MYC ‐driven inhibition of the glutamate‐cysteine ligase promotes glutathione depletion in liver cancer.. EMBO Rep. 2017, 18, 569-585, https://doi.org/10.15252/embr.201643068..
- Calvisi, D.F.; Simile, M.M.; Ladu, S.; Pellegrino, R.; De Murtas, V.; Pinna, F.; Tomasi, M.L.; Frau, M.; Virdis, P.; De Miglio, M.R.; et al.et al. Altered methionine metabolism and global DNA methylation in liver cancer: Relationship with genomic instability and prognosis. . Int. J. Cancer 2007, 121, 2410-2420, https://doi.org/10.1002/ijc.22940..
- Lee, D.; Xu, I.M.-J.; Chiu, D.K.-C.; Lai, R.K.-H.; Tse, A.P.-W.; Li, L.L.; Law, C.-T.; Tsang, F.H.-C.; Wei, L.L.; Chan, C.Y.-K.; et al.et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma.. J. Clin. Investig. 2017, 127, 1856-1872, https://doi.org/10.1172/jci90253..
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F.; Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer.. Nat. Commun. 2018, 9, 3164.
- Oh, B.-K.; Kim, H.; Park, H.-J.; Shim, Y.-H.; Choi, J.; Park, C.; Park, Y.N.; DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinico-pathological correlation.. Int. J. Mol. Med. 2007, 20, 65-73, https://doi.org/10.3892/ijmm.20.1.65..
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al.et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non‐histones and promotes tumorigenesis.. EMBO Rep. 2020, 22, e50967, https://doi.org/10.15252/embr.202050967..
- 67. Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al.et al. Histone Deacetylase HDAC8 Promotes Insulin Resistance and beta-Catenin Activation in NAFLD-Associated Hepatocellular Carcinoma. . Cancer Res. 2015, 75, 4803-4816.
- Jiang, H.; Cao, H.-J.; Ma, N.; Bao, W.-D.; Wang, J.-J.; Chen, T.-W.; Zhang, E.-B.; Yuan, Y.-M.; Ni, Q.-Z.; Zhang, F.-K.; et al.et al. Chromatin remodeling factor ARID2 suppresses hepatocellular carcinoma metastasis via DNMT1-Snail axis. . Proc. Natl. Acad. Sci. USA 2020, 117, 4770-4780, https://doi.org/10.1073/pnas.1914937117..
- Menk, A.V.; Scharping, N.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al.et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. . Cell Rep. 2018, 22, 1509-1521, https://doi.org/10.1016/j.celrep.2018.01.040..
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al.et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression.. Cell 2015, 162, 1229-1241, https://doi.org/10.1016/j.cell.2015.08.016..
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al.et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen.. Nat. Immunol. 2020, 21, 1022-1033, https://doi.org/10.1038/s41590-020-0725-2..
- Liu, P.-T.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al.et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. . Nat. Immunol. 2017, 18, 985-994, https://doi.org/10.1038/ni.3796..
- 99. Sinclair, L.V.; Howden, A.J.; Brenes, A.; Spinelli, L.; Hukelmann, J.L.; Macintyre, A.N.; Liu, X.; Thomson, S.; Taylor, P.M.; Rathmell, J.C.; et al.et al. Antigen receptor control of methionine metabolism in T cells.. eLife 2019, 12, 1-16, https://doi.org/10.7554/elife.44210..
- 102. Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Bruestle, A.; Itsumi, M.; et al.et al. Glutathione Primes T Cell Metabolism for Inflammation.. Immunity 2017, 46, 675-689, https://doi.org/10.1016/j.immuni.2017.03.019..
- 105. Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al.et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. . Immunity 2019, 51, 997-1011.e7, https://doi.org/10.1016/j.immuni.2019.11.009..
- 107. Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.-H.; et al.et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. . Cell Rep. 2019, 27, 2063-2074.e5, https://doi.org/10.1016/j.celrep.2019.04.022..
- 101. Hung, M.H.; Lee, J.S.; Ma, C.; Diggs, L.P.; Heinrich, S.; Chang, C.W.; Ma, L.; Forgues, M.; Budhu, A.; Chaisaing-mongkol, J.; et al.et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma.. Nat. Commun. 2021, 12, 1-15, https://doi.org/10.1038/s41467-021-21804-1..
- 148. Jühling, F.; Hamdane, N.; Crouchet, E.; Li, S.; El Saghire, H.; Mukherji, A.; Fujiwara, N.; A Oudot, M.; Thumann, C.; Saviano, A.; et al.et al. Targeting clinical epigenetic reprogramming for chemoprevention of metabolic and viral hepatocellular carcinoma. . Gut 2020, 70, 157-169, https://doi.org/10.1136/gutjnl-2019-318918..

