 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hirokazu Tsukahara | + 2162 word(s) | 2162 | 2021-10-11 08:25:21 | | | |
| 2 | Camila Xu | Meta information modification | 2162 | 2021-12-10 05:06:41 | | |
Video Upload Options
The respiratory system is continuously exposed to endogenous and exogenous oxidants. Chronic obstructive pulmonary disease (COPD) is characterized by chronic inflammation of the airways, leading to the destruction of lung parenchyma (emphysema) and declining pulmonary function. It is increasingly obvious that reactive oxygen species (ROS) and reactive nitrogen species (RNS) contribute to the progression and amplification of the inflammatory responses related to this disease. First, we described the association between cigarette smoking, the most representative exogenous oxidant, and COPD and then presented the multiple pathophysiological aspects of ROS and antioxidative defense systems in the development and progression of COPD. Second, the relationship between nitric oxide system (endothelial) dysfunction and oxidative stress has been discussed. Third, we have provided data on the use of these biomarkers in the pathogenetic mechanisms involved in COPD and its progression and presented an overview of oxidative stress biomarkers having clinical applications in respiratory medicine, including those in exhaled breath, as per recent observations. Finally, we explained the findings of recent clinical and experimental studies evaluating the efficacy of antioxidative interventions for COPD. Future breakthroughs in antioxidative therapy may provide a promising therapeutic strategy for the prevention and treatment of COPD.
1. Introduction
2. Cigarette Smoke and Oxidative Stress in COPD
3. Other Sources of Oxidative Stress in COPD
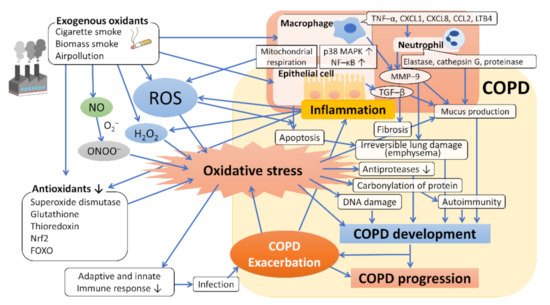
4. Oxidative Stress in the Development and Progression of COPD
References
- Chapman, K.R.; Mannino, D.M.; Soriano, J.B.; Vermeire, P.A.; Buist, A.S.; Thun, M.J.; Connell, C.; Jemal, A.; Lee, T.A.; Miravitlles, M.; et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 188–207.
- Soriano, J.B.; Abajobir, A.A.; Abate, K.H.; Abera, S.F.; Agrawal, A.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; Aichour, M.T.; Alam, K.; et al. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir. Med. 2017, 5, 691–706.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2021 Report). Available online: https://goldcopd.org/2021-gold-reports/ (accessed on 15 July 2021).
- Szalontai, K.; Gemes, N.; Furak, J.; Varga, T.; Neuperger, P.; Balog, J.A.; Puskas, L.G.; Szebeni, G.J. Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer. J. Clin. Med. 2021, 10, 2889.
- Taniguchi, A.; Miyahara, N.; Oda, N.; Morichika, D.; Ichihara, E.; Oze, I.; Tanimoto, Y.; Ichikawa, H.; Fujii, U.; Tanimoto, M.; et al. Protective Effects of Bisoprolol against Acute Exacerbation in Moderate-to-Severe Chronic Obstructive Pulmonary Disease. Acta Med. Okayama 2017, 71, 453–457.
- Oda, N.; Miyahara, N.; Ichikawa, H.; Tanimoto, Y.; Kajimoto, K.; Sakugawa, M.; Kawai, H.; Taniguchi, A.; Morichika, D.; Tanimoto, M.; et al. Long-term effects of beta-blocker use on lung function in Japanese patients with chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1119–1124.
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273.
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L205–L218.
- Fischer, B.M.; Voynow, J.A.; Ghio, A.J. COPD: Balancing oxidants and antioxidants. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 261–276.
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. 2017, 131, 1541–1558.
- Hou, H.H.; Cheng, S.L.; Chung, K.P.; Kuo, M.Y.; Yeh, C.C.; Chang, B.E.; Lu, H.H.; Wang, H.C.; Yu, C.J. Elastase induces lung epithelial cell autophagy through placental growth factor: A new insight of emphysema pathogenesis. Autophagy 2014, 10, 1509–1521.
- Rennard, S.I.; Vestbo, J. COPD: The dangerous underestimate of 15%. Lancet 2006, 367, 1216–1219.
- Rutgers, S.R.; Postma, D.S.; ten Hacken, N.H.; Kauffman, H.F.; van Der Mark, T.W.; Koëter, G.H.; Timens, W. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 2000, 55, 12–18.
- Kurimoto, E.; Miyahara, N.; Kanehiro, A.; Waseda, K.; Taniguchi, A.; Ikeda, G.; Koga, H.; Nishimori, H.; Tanimoto, Y.; Kataoka, M.; et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir. Res. 2013, 14, 5.
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27.
- Fujii, U.; Miyahara, N.; Taniguchi, A.; Waseda, K.; Morichika, D.; Kurimoto, E.; Koga, H.; Kataoka, M.; Gelfand, E.W.; Cua, D.J.; et al. IL-23 Is Essential for the Development of Elastase-Induced Pulmonary Inflammation and Emphysema. Am. J. Respir. Cell Mol. Biol. 2016, 55, 697–707.
- Waseda, K.; Miyahara, N.; Taniguchi, A.; Kurimoto, E.; Ikeda, G.; Koga, H.; Fujii, U.; Yamamoto, Y.; Gelfand, E.W.; Yamamoto, H.; et al. Emphysema requires the receptor for advanced glycation end-products triggering on structural cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 482–491.
- Morichika, D.; Miyahara, N.; Fujii, U.; Taniguchi, A.; Oda, N.; Senoo, S.; Kataoka, M.; Tanimoto, M.; Kakuta, H.; Kiura, K.; et al. A retinoid X receptor partial agonist attenuates pulmonary emphysema and airway inflammation. Respir. Res. 2019, 20, 2.
- Morichika, D.; Taniguchi, A.; Oda, N.; Fujii, U.; Senoo, S.; Itano, J.; Kanehiro, A.; Kitaguchi, Y.; Yasuo, M.; Hanaoka, M.; et al. Loss of IL-33 enhances elastase-induced and cigarette smoke extract-induced emphysema in mice. Respir. Res. 2021, 22, 150.
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544.
- Stämpfli, M.R.; Anderson, G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 2009, 9, 377–384.
- Pryor, W.A. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ. Health Perspect. 1997, 105 (Suppl. 4), 875–882.
- Churg, A.; Cosio, M.; Wright, J.L. Mechanisms of cigarette smoke-induced COPD: Insights from animal models. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L612–L631.
- MacNee, W. Oxidants and COPD. Curr. Drug Targets Inflamm. Allergy 2005, 4, 627–641.
- MacNee, W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 50–60.
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126.
- Pryor, W.A. Cigarette smoke and the involvement of free radical reactions in chemical carcinogenesis. Br. J. Cancer Suppl. 1987, 8, 19–23.
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57.
- Song, Q.; Chen, P.; Liu, X.M. The role of cigarette smoke-induced pulmonary vascular endothelial cell apoptosis in COPD. Respir. Res. 2021, 22, 39.
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379.
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127.
- Gensch, E.; Gallup, M.; Sucher, A.; Li, D.; Gebremichael, A.; Lemjabbar, H.; Mengistab, A.; Dasari, V.; Hotchkiss, J.; Harkema, J.; et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J. Biol. Chem. 2004, 279, 39085–39093.
- Di, Y.P.; Zhao, J.; Harper, R. Cigarette smoke induces MUC5AC protein expression through the activation of Sp1. J. Biol. Chem. 2012, 287, 27948–27958.
- Mebratu, Y.A.; Schwalm, K.; Smith, K.R.; Schuyler, M.; Tesfaigzi, Y. Cigarette smoke suppresses Bik to cause epithelial cell hyperplasia and mucous cell metaplasia. Am. J. Respir. Crit. Care Med. 2011, 183, 1531–1538.
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743.
- Siddharthan, T.; Gupte, A.; Barnes, P.J. Chronic Obstructive Pulmonary Disease Endotypes in Low- and Middle-Income Country Settings: Precision Medicine for All. Am. J. Respir. Crit. Care Med. 2020, 202, 171–172.
- Hagstad, S.; Backman, H.; Bjerg, A.; Ekerljung, L.; Ye, X.; Hedman, L.; Lindberg, A.; Torén, K.; Lötvall, J.; Rönmark, E.; et al. Prevalence and risk factors of COPD among never-smokers in two areas of Sweden—Occupational exposure to gas, dust or fumes is an important risk factor. Respir. Med. 2015, 109, 1439–1445.
- Thomsen, M.; Nordestgaard, B.G.; Vestbo, J.; Lange, P. Characteristics and outcomes of chronic obstructive pulmonary disease in never smokers in Denmark: A prospective population study. Lancet Respir. Med. 2013, 1, 543–550.
- Shin, S.; Bai, L.; Burnett, R.T.; Kwong, J.C.; Hystad, P.; van Donkelaar, A.; Lavigne, E.; Weichenthal, S.; Copes, R.; Martin, R.V.; et al. Air Pollution as a Risk Factor for Incident Chronic Obstructive Pulmonary Disease and Asthma. A 15-Year Population-based Cohort Study. Am. J. Respir. Crit. Care Med. 2021, 203, 1138–1148.
- Guo, C.; Zhang, Z.; Lau, A.K.H.; Lin, C.Q.; Chuang, Y.C.; Chan, J.; Jiang, W.K.; Tam, T.; Yeoh, E.K.; Chan, T.C.; et al. Effect of long-term exposure to fine particulate matter on lung function decline and risk of chronic obstructive pulmonary disease in Taiwan: A longitudinal, cohort study. Lancet Planet. Health 2018, 2, e114–e125.
- Andersen, Z.J.; Hvidberg, M.; Jensen, S.S.; Ketzel, M.; Loft, S.; Sørensen, M.; Tjønneland, A.; Overvad, K.; Raaschou-Nielsen, O. Chronic obstructive pulmonary disease and long-term exposure to traffic-related air pollution: A cohort study. Am. J. Respir. Crit. Care Med. 2011, 183, 455–461.
- Brassington, K.; Selemidis, S.; Bozinovski, S.; Vlahos, R. New frontiers in the treatment of comorbid cardiovascular disease in chronic obstructive pulmonary disease. Clin. Sci. 2019, 133, 885–904.
- McKelvey, M.C.; Brown, R.; Ryan, S.; Mall, M.A.; Weldon, S.; Taggart, C.C. Proteases, Mucus, and Mucosal Immunity in Chronic Lung Disease. Int. J. Mol. Sci. 2021, 22, 5018.
- Yao, H.; Arunachalam, G.; Hwang, J.W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576.
- Hwang, J.W.; Rajendrasozhan, S.; Yao, H.; Chung, S.; Sundar, I.K.; Huyck, H.L.; Pryhuber, G.S.; Kinnula, V.L.; Rahman, I. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J. Immunol. 2011, 187, 987–998.
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564.
- Mizumura, K.; Maruoka, S.; Shimizu, T.; Gon, Y. Role of Nrf2 in the pathogenesis of respiratory diseases. Respir. Investig. 2020, 58, 28–35.
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255.
- Barnes, P.J. The cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638.
- Kim, H.J.; Park, Y.D.; Moon, U.Y.; Kim, J.H.; Jeon, J.H.; Lee, J.G.; Bae, Y.S.; Yoon, J.H. The role of Nox4 in oxidative stress-induced MUC5AC overexpression in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 598–609.

