The respiratory system is continuously exposed to endogenous and exogenous oxidants. Chronic obstructive pulmonary disease (COPD) is characterized by chronic inflammation of the airways, leading to the destruction of lung parenchyma (emphysema) and declining pulmonary function. It is increasingly obvious that reactive oxygen species (ROS) and reactive nitrogen species (RNS) contribute to the progression and amplification of the inflammatory responses related to this disease. First, we described the association between cigarette smoking, the most representative exogenous oxidant, and COPD and then presented the multiple pathophysiological aspects of ROS and antioxidative defense systems in the development and progression of COPD. Second, the relationship between nitric oxide system (endothelial) dysfunction and oxidative stress has been discussed. Third, we have provided data on the use of these biomarkers in the pathogenetic mechanisms involved in COPD and its progression and presented an overview of oxidative stress biomarkers having clinical applications in respiratory medicine, including those in exhaled breath, as per recent observations. Finally, we explained the findings of recent clinical and experimental studies evaluating the efficacy of antioxidative interventions for COPD. Future breakthroughs in antioxidative therapy may provide a promising therapeutic strategy for the prevention and treatment of COPD.
1. Introduction
Globally, it is a leading cause of disability and death and exerts a great strain on healthcare resources [
1,
2]. Various studies on the development of new therapies for COPD and public health approaches are ongoing; however, no radical cure has been found thus far. Furthermore, the complications and quality of life associated with COPD have not been sufficiently improved, and >3 million people die from COPD each year [
3,
4].
The factors affecting the development and progression of COPD include genetic factors, age, sex, lung growth and development, exposure to particles, socioeconomic status, asthma and airway hyperresponsiveness, chronic bronchitis, and infections [
3,
5,
6]. Among the related particles, cigarette smoke is the most commonly encountered risk factor for COPD [
3].
However, the detailed mechanisms by which cigarette smoking causes COPD have not been elucidated. Protease–antiprotease imbalance, neutrophilic airway inflammation induced by immune responses, oxidative stress [
7,
8,
9,
10], and apoptosis [
11] have been reported to be involved in the development of COPD. However, the fact that only a relatively small percentage of smokers develop COPD [
12] and that airway inflammation persists, and the disease progresses even after smoking cessation [
13] suggests that other factors, including host genetic factors, are greatly involved in the development and progression of this disease.
COPD is associated with chronic inflammation characterized by an increased number of alveolar macrophages, neutrophils, T lymphocytes (predominantly Tc1, Th1, and Th17 cells), and innate lymphoid cells recruited from the circulation [
14,
15,
16]. These inflammatory cells and structural cells, including epithelial and endothelial cells and fibroblasts, secrete several pro-inflammatory mediators, including cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-8; chemokines, such as C-X-C motif ligand (CXCL) 1; CXCL8, C-C motif ligand (CCL)-2; growth factors, such as transforming growth factor (TGF)-β; and lipid mediators, such as leukotriene (LT) B
4 [
15,
17,
18,
19].
Inflammation in COPD is thought to be closely related to oxidative stress [
7,
8,
9,
10,
15]. In patients with COPD, oxidative stress occurs because endogenous antioxidant defenses are genetically impaired and/or overwhelmed by the presence of reactive oxygen species (ROS) [
7]. One of the sources of ROS is environmental sources, represented by cigarette smoke, and is called exogenous ROS. Conversely, cellular sources, including inflammatory and structural cells, produce ROS upon activation of xanthine oxidase (XO) and nicotine adenine disphosphonucleotide (NADPH) oxidase and/or by mitochondria [
7] and are called endogenous ROS. In patients with COPD, oxidative stress occurs after long-term exposure to environmental cigarette smoke and combustion products of biomass fuels, as well as due to a variety of immune and inflammatory stimuli in the airways [
20].
2. Cigarette Smoke and Oxidative Stress in COPD
The leading environmental risk factor for COPD is cigarette smoking [
3]. Cigarette smoke comprises >4500 components in its gaseous and particulate phases. These compounds include direct carcinogens (methylcholanthrene, benzo-α-pyrenes, and acrolein), toxins (carbon monoxide, nicotine, ammonia, acetone, and hydroquinone), reactive solids with chemically catalytic surfaces [
22], and numerous oxidant compounds that have been identified among the 4000–7000 constituents of cigarette smoke [
23]. Furthermore, cigarette smoke contains a high concentration of ROS [
24] and is considered to be associated with higher levels of oxidative stress in cigarette smokers [
25,
26]. A single puff of cigarette smoke contains 10
17 free radicals in the tar phase and 10
15 free radicals in the gas phase [
27,
28]. For example, superoxide (O
2−), epoxides, peroxides, nitric oxide (NO), nitrogen dioxide (NO
2), peroxynitrite (ONOO
−), and peroxynitrate (O
2NOO
−) are present in the gas phase [
9].
In addition to inducing ROS, cigarette smoke affects the endogenous oxidant/antioxidant balance and modifies the production of proteins and action of enzymes by epigenetic modulation mechanisms in cells, such as bronchoalveolar epithelial cells and macrophages. Cigarette smoke promotes the activation of inflammatory cells through the activation of transcription factors, such as nuclear factor-κB (NF-κB), p38 mitogen-activated protein kinase (MAPK), and posttranslational modification of histone deacetylase (HDAC) in macrophages [
8,
29]. These responses trigger the cells to release proinflammatory cytokines, to recruit more neutrophils, macrophages, and dendritic cells and to exacerbate the inflammatory process [
8]. Newly recruited inflammatory cells continue the inflammatory process through phagocytosis, cytokine secretion, and surface antigen formation, mediating the innate and adaptive immune responses, while inflammatory and structural cells generate endogenous ROS [
8]. The burden of oxidants/ROS in the respiratory tract, through the prooxidant/antioxidant imbalance induced by the activation of inflammatory cells and gene regulatory mechanism described below, results in lung cell damage and induces pulmonary vascular endothelial cell apoptosis [
8,
30], which delays the resolution of inflammation by compromising the phagocytic ability of alveolar macrophages, leading to necrosis and emphysema [
8].
The host’s antioxidant capacity is attenuated by the coordination of cigarette smoking itself, ROS, and various mediators of the associated inflammation, resulting in enhanced oxidative stress [
20]. In addition, nitrosative stress is caused by cigarette smoke, which is a source of reactive nitrogen species (RNS). Large amounts of NO, which is abundant in cigarette smoke and is also generated by inducible NO synthase (NOS
2) in inflammatory cells, reacts with oxygen (O
2) and O
2− to produce highly oxidizing NO
2 and ONOO
−, respectively [
20,
31,
32].
Furthermore, cigarette smoke is considered to induce apoptosis of pulmonary vascular endothelial cells via epigenetic mechanisms by modulating various regulatory mechanisms, such as deoxyribonucleic acid (DNA) methylation, ribonucleic acid (RNA) methylation, histone modification, microRNA (miRNA), exosomes, and noncoding RNA [
30].
Cigarette smoke upregulates mucin 5AC (MUC5AC) expression via activation of activator protein 1 or specificity protein 1 [
33,
34]. In addition, cigarette smoke constituents, including aldehydes (e.g., acrolein) and hydrogen peroxide (H
2O
2), also upregulate mucin gene expression [
9]. Cigarette smoke also prevents the apoptosis of airway epithelial cells by suppressing Bik, a proapoptotic molecule, thereby increasing epithelial cell hyperplasia [
35].
Long-term exposure to cigarette smoke impairs phagocytosis and antigen-presentation functions of neutrophils and macrophages, which predispose patients to respiratory tract infections. Chronic inflammation and bacterial colonization in the lower respiratory tract, combined with compromised host defenses, could explain the progression of COPD even after smoking cessation [
8].
3. Other Sources of Oxidative Stress in COPD
It has been reported that approximately 25–45% of patients with COPD have never smoked; therefore, we cannot neglect other risk factors of COPD that can induce oxidative stress leading to COPD [
36]. Exposure to biomass has a 2.5–3 times higher risk of airflow limitation; especially, the use of biomass fuel for cooking in low-and-middle income countries is assumed to be one of the important risk factors for COPD [
37]. Occupational exposure to dust, gas, and fumes was also significantly associated with an increased risk of COPD [
38,
39].
The association between ambient air pollution and the incidence of COPD has also been reported. Ozone induces bronchial inflammation through oxidative injury. It has been reported that ozone level is associated with COPD incidence [
40]. Associations of other ambient air pollutants, such as particulate matter 2.5 and NO
2, with the incidence of COPD have also been reported [
41,
42].
4. Oxidative Stress in the Development and Progression of COPD
Enhancement of oxidative stress is thought to be caused by the coordination of exogenous oxidants, such as cigarette smoke, biomass smoke, and air pollution; endogenous ROS, such as O
2− and H
2O
2; and reduction of antioxidants, such as superoxide dismutase (SOD), glutathione (GSH), and nuclear factor erythroid 2-related factor 2 (Nrf2) [
20]. Enhanced oxidative stress triggers various reactions, including enhancing inflammation through the activation of transcription factors (such as NF-κB and p38 MAPK), producing autoantibodies to activate autoimmunity and lowering antiproteases to promote emphysema [
20].
Macrophages play a key role in orchestrating chronic inflammation in patients with COPD and can be activated by cigarette smoke extract to release inflammatory mediators, including TNF-α, CXCL1, CXCL8, CCL2, LTB
4, and ROS [
15]. Under the direction of these chemotactic mediators, neutrophils migrate into the respiratory tract and secrete serine proteases, including neutrophil elastase, cathepsin G, and proteinase-3, which damage tissues and stimulate mucus secretion from submucosal glands and goblet cells. Neutrophils also secrete matrix metalloproteinase (MMP)-8 and MMP-9, which contribute to alveolar destruction and development of emphysema [
15,
43,
44].
Decreased antioxidants also contribute significantly to increased oxidative stress. Antioxidants, such as SOD, GSH, thioredoxin, and Nrf2, are considered to play very important roles in the lungs, which are continuously exposed to the external environment. Expression of extracellular SOD3 polymorphisms is reduced around small airways in patients with COPD [
45], and the transcription factors that regulate multiple antioxidant genes, Nrf2 and FOXO3a (Forkhead box O3a), are reduced in lungs with COPD [
46,
47]. Nrf2 is a transcription factor that is involved in protection against oxidative damage by regulating the expression of genes, and its endogenous inhibitor, Kelch-like ECH-associated protein 1, prevents binding to the antioxidant response element [
48]. Moreover, disruption of Nrf2 causes early onset and severe emphysema, and Nrf2 activator, 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl] imidazole, attenuates cigarette smoke-induced emphysema and alveolar/cardiac dysfunction in mice [
49]. Even after cessation of particle exposure, COPD exacerbation continues by reducing the host antioxidant capacity, and oxidative stress is sustained after amelioration of exacerbations due to continuous production of ROS from endogenous sources, similar to the continuation of oxidative stress after smoking cessation [
7].
Over 50 cytokines and chemokines are released in the lungs of patients with COPD and contribute to increased inflammation [
50]. Many of the intracellular signaling pathways that trigger and/or drive the release of these inflammatory mediators are sensitive to oxidative stress due to the incorporation of redox-sensitive molecular targets, such as NF-κB, Ras/Rac, Jun-N-terminal kinase, p38 MAPK, and protein tyrosine phosphatases [
20]. Oxidative stress activates NF-κB and TGF-β signaling pathways in airway epithelial cells and macrophages, which induce oxidative stress [
20] and are involved in small airway fibrosis. As a result of the activation of these intracellular signaling pathways triggered and regulated by oxidative stress, the airway lumen of patients with COPD is characterized by an increased number of neutrophils, macrophages, T lymphocytes, and B lymphocytes. This inflammation is more amplified in patients with COPD than in smokers without COPD and increases further during acute exacerbations or when precipitated by bacterial/viral infection [
15].
Oxidative stress also regulates the expression of mucin genes, such as MUC5AC and mucous cell metaplasia [
9]. Exposure to H
2O
2, which is a constituent of cigarette smoke and an endogenous oxidant, upregulates MUC5AC mRNA expression via an NADPH oxidoreductase 4-dependent mechanism [
51].
In severe COPD, autoantibodies against epithelial and endothelial cells are generated, causing autoimmunity. Oxidative stress is considered to cause carbonylation of proteins, which creates neoantigens and induces the production of autoantibodies [
20]. Furthermore, oxidative stress directly damages the DNA. Apurinic/apyradymic sites, which are common DNA lesions in the repair of oxidized bases, are increased in the lungs of smokers without COPD, reflecting active DNA repair, whereas they are reduced in the lungs of patients with COPD, indicating defective DNA repair [
20]. Many of these responses to oxidative stress may eventually be involved in the development and progression of COPD; moreover, acute exacerbation causes a further increase in oxidative stress, which also leads to the progression of COPD. A summary of these points is presented in
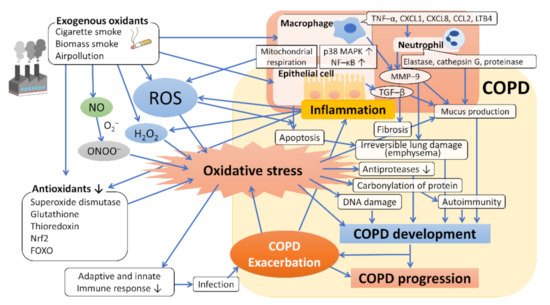
Figure 1.
Figure 1. Oxidative stress and the mechanism underlying the development and progression of COPD. Exogenous oxidants such as cigarette smoke contain ROS, which act to promote oxidative stress and induce inflammation of macrophages and bronchoalveolar epithelial cells by activating transcription factors such as NF–κB, which leads to further ROS generation (endogenous ROS). The host’s antioxidants, such as superoxide dismutase, glutathione, thioredoxin, and Nrf2, are attenuated by the coordination of exogenous oxidants and various mediators of the associated inflammation, resulting in enhanced oxidative stress. Oxidative stress regulates the expression of the mucin gene to increase mucin secretion, inducing DNA damage and causing autoimmunity through carbonylation of proteins, and reduces antiprotease activity, leading to the development and progression of emphysema and fibrosis. Abbreviations: ROS, reactive oxygen species; H2O2, hydrogen peroxide; NO, nitric oxide; O2–, superoxide anion; ONOO–, peroxynitrite; NF–κB, nuclear factor–κB; p38 MAPK, p38 mitogenactivated protein kinase; Nrf2, nuclear factor erythroid 2–related factor 2; FOXO, forkhead box O; TNF–α, tumor necrosis factor–α; LTB4, leukotriene B4; GM–CSF, granulocyte macrophage colony–stimulating factor; TGF–β, transforming growth factor–β; MMP–9, metalloproteinase–9.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10101537