 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maya Saleh | + 2845 word(s) | 2845 | 2021-11-01 09:27:24 | | | |
| 2 | Lindsay Dong | + 597 word(s) | 3442 | 2021-12-08 10:49:45 | | |
Video Upload Options
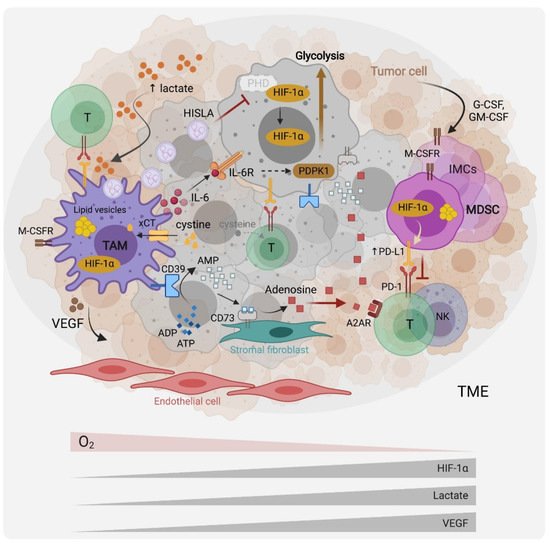
In tumors, myeloid cells, including tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), represent a predominant immune population with significant heterogeneity.
1. Introduction
2. The Role of Hypoxia, Aerobic Glycolysis and Lactate in the Recruitment and Functions of TAMs and MDSCs
3. Amino Acids, Metabolism Controls, Myeloid Cell Phenotypes and Functions in the TME
3.1. Glutamine
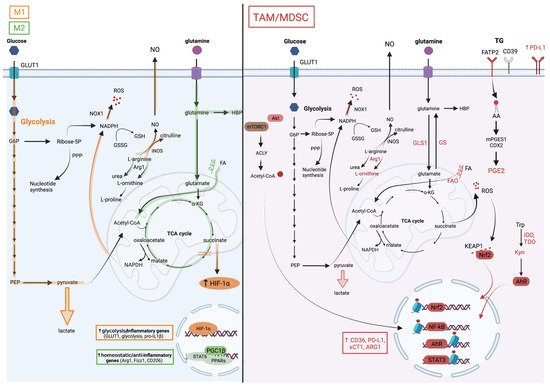
Glutamine is a key metabolite required for several cellular processes, such as nucleotide synthesis, amino acid production and glycosylation. Glutamine is provided from exogenous intake or produced by glutamine synthetase (GS, also known as Glutamate-Ammonia Ligase [GLUL]) from glutamate and ammonia (Figure 2). Several studies have reinforced the dependency of pro-tumoral myeloid cells on glutamine metabolism.
On one hand, the genetic or pharmacological inhibition of GS in macrophages has been shown to rewire their metabolism towards a pro-inflammatory phenotype impacting the activity of macrophages in cancer metastasis in vivo. For instance, using the M1/M2 culture system, Palmieri et al. demonstrated that the inhibition of the GS-modulated macrophage metabolism and function towards an M1 profile notably led to succinate accumulation, HIF-1α stabilization, and to a decreased ability of IL-10-treated macrophages to suppress T cell activity, and to promote the endothelial capillary network formation [27].
On the other hand, the interference with glutaminolysis by inhibiting glutaminase (GLS1) activity was similarly effective in blunting the immunosuppressive phenotype of macrophages, tumor-associated immature myeloid cells and MDSCs. Such a treatment increased the efficacy of the immune checkpoint blockade, highlighting the potential for inhibiting myeloid cell metabolic checkpoints for cancer immunotherapy. Initially, Liu et al. used in vitro M1/M2 cultures to demonstrate that glutaminolysis, leading to the generation of αKG and thus a high αKG/succinate ratio, promoted the metabolic and epigenetic upregulation of M2 genes via the αKG-dependent histone demethylase Jumonji d3 (jmjd3) [28]. In contrast, in M1 macrophages, αKG suppressed the NF-κB pathway by inhibiting IKK through PHD-dependent proline hydroxylation, which led to impaired proinflammatory responses [28]. Glutaminolysis is not only implicated in macrophage polarization, but also plays a role in the differentiation and function of immunosuppressive myeloid cells. Both Wu et al. [29] and Sun et al. [30] demonstrated the presence of hematopoietic progenitor cells (HPC) in cancers that give rise to immature myeloid cells (IMC) and MDSCs.
3.2. Tryptophan
3.3. Arginine
3.4. Cystine, Glutamate and Oxidative Stress
4. Lipids and Cholesterol Metabolism Shape Immunosuppressive Myeloid Cells in the TME
5. Myelosuppressive Effects of Adenosine
6. Wnt Ligands Potentiate Myeloid Cells—Cancer Cells Metabolic Crosstalk
7. Conclusions and Perspectives
References
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.-M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363, eaau0964.
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904.
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744.
- Werno, C.; Menrad, H.; Weigert, A.; Dehne, N.; Goerdt, S.; Schledzewski, K.; Kzhyshkowska, J.; Brüne, B. Knockout of HIF-1 in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 2010, 31, 1863–1872.
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714.
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709.
- Alexander, R.K.; Liou, Y.-H.; Knudsen, N.H.; Starost, K.A.; Xu, C.; Hyde, A.L.; Liu, S.; Jacobi, D.; Liao, N.-S.; Lee, C.-H. Bmal1 integrates mitochondrial metabolism and macrophage activation. eLife 2020, 9, 54090.
- De-Brito, N.M.; Duncan-Moretti, J.; Da-Costa, H.C.; Saldanha-Gama, R.; Paula-Neto, H.A.; Dorighello, G.; Simões, R.L.; Barja-Fidalgo, C. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim. Biophys. Acta BBA Bioenerg. 2020, 1867, 118604.
- Wu, J.-Y.; Huang, T.-W.; Hsieh, Y.-T.; Wang, Y.-F.; Yen, C.-C.; Lee, G.-L.; Yeh, C.-C.; Peng, Y.-J.; Kuo, Y.-Y.; Wen, H.-T.; et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell 2020, 77, 213–227.
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453.
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790.
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976.
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371.
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103.
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G.; et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644.
- Katholnig, K.; Schütz, B.; Fritsch, S.D.; Schörghofer, D.; Linke, M.; Sukhbaatar, N.; Matschinger, J.M.; Unterleuthner, D.; Hirtl, M.; Lang, M.; et al. Inactivation of mTORC2 in macrophages is a signature of colorectal cancer that promotes tumorigenesis. JCI Insight 2019, 4, 24.
- Ding, C.; Sun, X.; Wu, C.; Hu, X.; Zhang, H.-G.; Yan, J. Tumor microenvironment modulates immunological outcomes of myeloid cells with mTORC1 disruption. J. Immunol. 2019, 202, 1623–1634.
- Collins, S.L.; Oh, M.-H.; Sun, I.-H.; Chan-Li, Y.; Zhao, L.; Powell, J.D.; Horton, M.R. mTORC1 signaling regulates proinflammatory macrophage function and metabolism. J. Immunol. 2021, 207, 913–922.
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 2016, 5, e11612.
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver′s seat. Nat. Rev. Cancer 2017, 17, 577–593.
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939.
- Liu, N.; Luo, J.; Kuang, D.; Xu, S.; Duan, Y.; Xia, Y.; Wei, Z.; Xie, X.; Yin, B.; Chen, F.; et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α–mediated tumor progression. J. Clin. Investig. 2019, 129, 631–646.
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563.
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483.
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; Heiland, D.H.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110.
- Liu, Z.; Xu, J.; Ma, Q.; Zhang, X.; Yang, Q.; Wang, L.; Cao, Y.; Xu, Z.; Tawfik, A.; Sun, Y.; et al. Glycolysis links reciprocal activation of myeloid cells and endothelial cells in the retinal angiogenic niche. Sci. Transl. Med. 2020, 12, eaay1371.
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017, 20, 1654–1666.
- Liu, P.-T.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994.
- Wu, W.-C.; Sun, H.-W.; Chen, J.; Ouyang, H.-Y.; Yu, X.-J.; Chen, H.-T.; Shuang, Z.-Y.; Shi, M.; Wang, Z.; Zheng, L. Immunosuppressive immature myeloid cell generation is controlled by glutamine metabolism in human cancer. Cancer Immunol. Res. 2019, 7, 1605–1618.
- Sun, H.-W.; Wu, W.-C.; Chen, H.-T.; Xu, Y.-T.; Yang, Y.-Y.; Chen, J.; Yu, X.-J.; Wang, Z.; Shuang, Z.-Y.; Zheng, L. Glutamine deprivation promotes the generation and mobilization of MDSCs by enhancing expression of G-CSF and GM-CSF. Front. Immunol. 2021, 11, 616367.
- Zhao, Q.; Kuang, D.-M.; Wu, Y.; Xiao, X.; Li, X.-F.; Li, T.-J.; Zheng, L. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J. Immunol. 2012, 188, 1117–1124.
- Su, S.; Zhao, J.; Xing, Y.; Zhang, X.; Liu, J.; Ouyang, Q.; Chen, J.; Su, F.; Liu, Q.; Song, E. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 2018, 175, 442–457.e23.
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203.
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.-C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Author correction: Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 1533.
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698.
- Brentville, V.A.; Metheringham, R.L.; Daniels, I.; Atabani, S.; Symonds, P.; Cook, K.W.; Vankemmelbeke, M.; Choudhury, R.; Vaghela, P.; Gijon, M.; et al. Combination vaccine based on citrullinated vimentin and enolase peptides induces potent CD4-mediated anti-tumor responses. J. Immunother. Cancer 2020, 8, e000560.
- Mills, C.D.; Shearer, J.; Evans, R.; Caldwell, M.D. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J. Immunol. 1992, 149, 2709–2714.
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849.
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77.
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2017, 167, 457–470.
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24.
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O′Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855.
- Yan, D.; Yang, Q.; Shi, M.; Zhong, L.; Wu, C.; Meng, T.; Yin, H.; Zhou, J. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur. J. Immunol. 2013, 43, 2943–2955.
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137.
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255.
- Cekic, C.; Day, Y.-J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259.
- Del Barrio, I.M.; Penski, C.; Schlahsa, L.; Stein, R.G.; Diessner, J.; Wöckel, A.; Dietl, J.; Lutz, M.B.; Mittelbronn, M.; Wischhusen, J.; et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages—A self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J. Immunother. Cancer 2016, 4, 49.
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21.
- Cosin-Roger, J.; Ortiz-Masià, M.D.; Barrachina, M.D. Macrophages as an emerging source of Wnt ligands: Relevance in mucosal integrity. Front. Immunol. 2019, 10, 2297.
- Yang, Y.; Ye, Y.C.; Chen, Y.; Zhao, J.L.; Gao, C.C.; Han, H.; Liu, W.C.; Qin, H.Y. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 793.

