2. The Role of Hypoxia, Aerobic Glycolysis and Lactate in the Recruitment and Functions of TAMs and MDSCs
Hypoxia is an environmental factor with a great impact on the regulation of myeloid cell profiles within the TME. In hypoxic conditions, hypoxia-induced factors (HIF)-1
α and HIF-2
α accumulate and engage several signaling pathways that converge on phenotypic and functional changes in myeloid cells. In 2010, Werno C et al. examined the role of HIF-1
α in macrophage tumor infiltration. They demonstrated that in a coculture in vitro system, a loss of HIF-1
α in macrophages derived from cd11b
+ splenocytes did not impact the capacity to infiltrate tumor spheroids
[4][9]. Imtiyaz et al. generated myeloid-specific HIF-2
α knockout mice and demonstrated that HIF-2
α was required for TAM infiltration, potentially by upregulating the expression of CXCR4 and M-CSFR
[5][10]. A follow-up study by Casazza A et al. reported the context-dependent role of HIF-2
α in the infiltration of TAMs, particularly in the hypoxic areas of the tumor. Although the initial attraction of TAMs to tumors is mediated by hypoxia-induced Semaphorin 3A and Neuropilin-1 (Nrp1) signaling, these cells then become “entrapped” via a secondary suppression of Nrp1 by HIF-2
α. In such a hypoxic environment, the macrophages acquire immunosuppressive and pro-angiogenic properties that promote tumor growth. In contrast, the forced retainment of TAMs in normoxic areas, through the genetic deletion of
Nrp1, results in their differentiation into inflammatory anti-tumoral cytotoxic macrophages
[6][11]. This work provided early evidence that the heterogeneity of TAMs depended on their localization with respect to an oxygen gradient.
In contrast to the M1/M2 immunometabolism paradigm, more recent studies have shown that immunosuppressive TAMs and MDSCs are highly glycolytic, and that aerobic glycolysis, HIF-1
α activity and succinate accumulation in the TME, promote their immunosuppressive phenotypes and functions. For instance, Alexander et al. demonstrated that the myeloid-specific deletion of the circadian clock regulator
Bmal1, led to impaired macrophage mitochondrial metabolism, the accumulation of mitochondrial reactive oxygen species (mROS) and succinate, HIF-1
α stabilization and enhanced aerobic glycolysis. Such an aberrant HIF-1
α activation in TAMs promoted the TAM-pro-tumoral function and tumor development
[7][15]. Another direct evidence of the high glycolytic activity of TAMs came from work by de-Brito et al., who showed that blocking glycolysis with 2-deoxy-glucose (2-DG) in macrophages derived from human monocytes and cultured in tumor-cell-line conditioned media diminished the production of M2 markers
[8][16]. Extracellular succinate further contributes to the migration of macrophages into the tumor site and their differentiation into tumor-promoting cells, by engaging its receptor SUCNR1 on the macrophage-cell surface, which drives the PI3K-HIF-1
α pathway
[9][17]. In contrast, extracellular succinate stimulates SUCNR1 on the tumor-cell surface furthering tumor proliferation and metastasis
[9][17]. HIF-1
α also promotes the differentiation of tumor MDSCs into more potent suppressors of T cell activity
[10][18]. A potential mechanism was proposed by Noman et al., who identified the immune checkpoint PD-L1 as a transcriptional target of HIF-1
α [11][19] (
Figure 1). Collectively, HIF-1
α potentiates aerobic glycolysis and pro-inflammatory cytokine production in an inflammatory setting. In the TME, HIF proteins drive the pro-tumoral activities of TAMs and the differentiation of MDSC into potent suppressors of anti-tumor immunity.
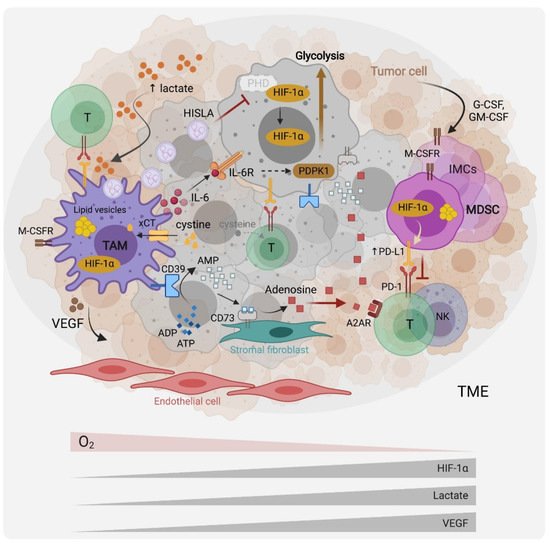
Figure 1. Myeloid cells metabolic interactions with other cells in the tumor microenvironment. Tumor cells produce G-CSF and GM-CSF that recruit myeloid cells, including immature myeloid cells (IMC), myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) to the TME. Hypoxia, which results in the stabilization of the hypoxia-induced factor (HIF)-1a, the tumor cell’s upregulation of aerobic glycolysis (the Warburg effect), and subsequent TME lactate accumulation and acidification, modulate myeloid cells towards pro-tumoral and immunosuppressive effectors. Through the expression of cytokines such as interleukin (IL)-6, immune checkpoints ligands such as Programmed Death Ligand 1 (PD-L1), and growth factors such as Vascular Endothelial Growth Factor (VEGF), myeloid cells promote tumor-cell survival, immune evasion and angiogenesis. Recently, TAMs were reported to promote the Warburg effect via IL-6 signaling as well as through vesicles containing a HIF-1a-stabilizing lncRNA (HISLA). Through the cystine transporter xCT, MDSCs and TAMs deplete the TME of cysteine, which is necessary for T cell effector functions. They further suppress T cell activity by upregulating the expression of the ectonucleotidases CD39 and CD73 at their surface, leading to adenosine production and immunosuppressive signaling via the adenosine receptor (A2AR). TAMs and MDSCs accumulate lipid droplets in cytosolic vesicles and appear to rely on lipid metabolism and fatty acid oxidation for their pro-tumoral functions.
Besides HIF proteins, the mechanistic target of rapamycin (mTOR) exerts important functions in myeloid-cell migration, polarization and function. The mTOR exists in one of two complexes, mTOR complex (mTORC)1 or mTORC2, by associating with either Raptor or Rictor, respectively. Moreover, mTOR signaling regulates a broad set of basic cellular and metabolic processes, including translation, cell growth and proliferation
[12][13][20,21], and has established roles in immunoregulation, acting in both innate and adaptive immune cells
[14][22]. The mTOR activation in cancer cells promotes tumorigenesis through various mechanisms including the recruitment of MDSCs, by inducing the production of the myelopoiesis and mobilizing cytokine G-CSF
[15][23]. To elucidate the specific contributions of the two mTOR complexes, mice with the myeloid-specific deletion of either
Raptor or
Rictor were generated. In the latter, mTORC2 was established as a regulator of M2 polarization and a negative regulator of pro-inflammatory macrophage differentiation
[16][24]. Consistently, mice with the myeloid-specific deletion of mTORC2 were more susceptible to colitis-associated colorectal cancer (CAC) exhibiting an enhanced production of inflammatory mediators, including SPP1/osteopontin
[16][24]. Notably, the role of mTORC1 in TAM differentiation has been controversial; Ding et al. used a subcutaneous transplantable Lewis lung carcinoma (LLC) mouse model and showed that the loss of Raptor in TAMs did not impact primary tumor growth, but enhanced lung metastasis by promoting the expansion of metastasis-associated macrophages
[17][25]. In contrast, Collins et al. recently reported that mTORC1 controlled M2 polarization
[18][26]. Despite impaired glycolysis, the myeloid-specific deletion of mTORC1 led to enhanced pro-inflammatory functions in vitro and in vivo, due to the enhanced histone acetylation downstream of the inhibited sirtuins
[18][26]. This is in line with previous work by Horng and coworkers, who identified the Akt-mTORC1 pathway as a regulator of ATP-citrate lyase (ACLY) that synthesizes cytosolic acetyl CoA and triggers M2 gene induction through histone acetylation
[19][27] (
Figure 2).
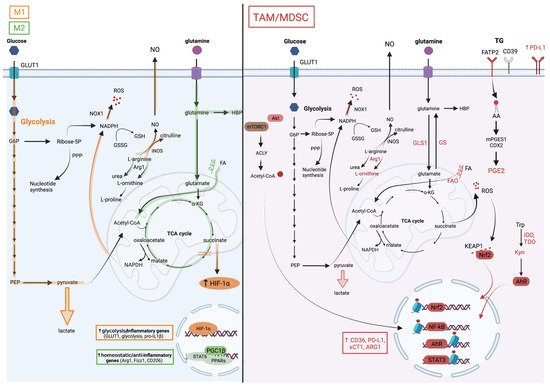
Figure 2. Metabolic pathways associated with TAM and MDSC pro-tumoral and myelosuppressive functions. The M1/M2 paradigm established from an in vitro macrophage culture system distinguishes two extreme macrophage metabolic phenotypes. (On the left), the metabolic pathways in orange are upregulated in M1 macrophages and those in green in M2 macrophages. M1 macrophages are highly glycolytic which is a consequence of HIF-1a stabilization in response to succinate accumulation due to breaks in the TCA cycle. Besides inducing the expression of glycolytic enzymes, HIF-1a also drives the production of pro-inflammatory factors such as IL-1b. In contrast, M2 macrophages favor glutamine consumption, upregulate the hexosamine biosynthetic pathway (HBP), and rely on fatty acid oxidation (FAO) for their energetic needs. STAT6 downstream of IL-4 signaling and PPARg with its co-activator PGC1b are key for their differentiation. In contrast to this simplified system, the TME complexity results in a marked myeloid cell heterogeneity with M1/M2 mixed profiles and divergent metabolic characteristics. (On the right), the cellular metabolic pathways upregulated in TAMs and MDSCs are illustrated. These cells are highly glycolytic but are dependent on glutamine and lipid consumption for their pro-tumoral functions. Despite their heightened aerobic glycolysis, they upregulate M2-like genes through the accumulation of acetyl coA, which is downstream of the AKT/mTOR-dependent activation of ATP Citrate Lyase (ACLY), and histone acetylation. Histone lactylation, which was reported to occur in M1 macrophages at later stages of activation and proposed as a mechanism to terminate the inflammatory response, might regulate TAM functions, but this remains to be tested. The heightened mitochondrial respiration in tumor-associated myeloid cells leads to the elevated production of reactive oxygen species (ROS). To withhold oxidative stress, they activate the transcription factor NRF2, which induces the expression of anti-oxidative genes and of the cystine transporter xCT1, among others. Myeloid cells in the TME upregulate triglycerides (TG) uptake, for instance through fatty acid transport protein (FATP)2, as reported in granulocytic 18 MDSCs, lipid accumulation in vesicles, lipolysis and FAO. Consequently, they also produce inflammatory and immunosuppressive lipid mediators such as the prostaglandin PGE2. Furthermore, they metabolize arginine and tryptophan into metabolites that favor tumor growth, including L-ornithine and L-kynurenine (Kyn). The latter is a ligand of aryl hydrocarbon receptor (AHR), which promotes myelosuppressive functions in the TME via transcriptional activity.
The key metabolic features of the TME include hypoxia, acidosis and lactate accumulation
[20][31], which collectively contribute to myeloid cell infiltration and their differentiation into pro-tumoral immunosuppressive effectors. Indeed, hypoxia and lactate gradients have been shown to act as TME morphogens, eliciting the progressive differentiation of TAMs into pro-angiogenic orchestrators
[21][32], partly through the HIF-induced production of vascular endothelial growth factor (VEGF)
[22][33] (
Figure 1). Such positional information might organize tumors into structured entities akin to the morphogenic regulation of embryonic tissue development. Mechanistically, lactate has been shown to activate mTORC1, resulting in the suppression of ATP6V0d2, a vacuolar ATPase involved in HIF-2
α lysosomal degradation. Such a lactate-driven link between mTORC1 activation and the stabilization of HIF-2
α favors for pro-tumoral macrophage differentiation
[22][33]. In addition, lactate rewires the macrophage metabolism into M2 cells, independently of IL-4/IL-13 signaling
[23][34].
In the cancer setting, TAMs often express an M1/M2 mixed phenotype
[24][37], as reaffirmed recently by single-cell approaches in several cancers, including glioblastoma
[25][38]. This has also been observed in non-cancerous settings in vivo. For example, using a retinopathy mouse model, Liu et al. showed that retinal microglia and macrophages adjacent to endothelial cells (EC) are highly glycolytic, pro-angiogenic and express both M1 and M2 markers, in response to the elevated lactate levels released by EC cells
[26][39]. Hyper-glycolytic macrophages accumulate acetyl-coA that epigenetically drives M2 pro-angiogenic factors, which in turn promote EC sprouting and the establishment of a pathological vascular niche
[26][39]. Two recent studies have further illustrated the metabolic crosstalk among cells in the TME favoring tumor growth.
3. Amino Acids, Metabolism Controls, Myeloid Cell Phenotypes and Functions in the TME
3.1. Glutamine
Glutamine is a key metabolite required for several cellular processes, such as nucleotide synthesis, amino acid production and glycosylation. Glutamine is provided from exogenous intake or produced by glutamine synthetase (GS, also known as Glutamate-Ammonia Ligase [GLUL]) from glutamate and ammonia (Figure 2). Several studies have reinforced the dependency of pro-tumoral myeloid cells on glutamine metabolism.
On one hand, the genetic or pharmacological inhibition of GS in macrophages has been shown to rewire their metabolism towards a pro-inflammatory phenotype impacting the activity of macrophages in cancer metastasis in vivo. For instance, using the M1/M2 culture system, Palmieri et al. demonstrated that the inhibition of the GS-modulated macrophage metabolism and function towards an M1 profile notably led to succinate accumulation, HIF-1α stabilization, and to a decreased ability of IL-10-treated macrophages to suppress T cell activity, and to promote the endothelial capillary network formation [27][42].
On the other hand, the interference with glutaminolysis by inhibiting glutaminase (GLS1) activity was similarly effective in blunting the immunosuppressive phenotype of macrophages, tumor-associated immature myeloid cells and MDSCs. Such a treatment increased the efficacy of the immune checkpoint blockade, highlighting the potential for inhibiting myeloid cell metabolic checkpoints for cancer immunotherapy. Initially, Liu et al. used in vitro M1/M2 cultures to demonstrate that glutaminolysis, leading to the generation of αKG and thus a high αKG/succinate ratio, promoted the metabolic and epigenetic upregulation of M2 genes via the αKG-dependent histone demethylase Jumonji d3 (jmjd3) [28][44]. In contrast, in M1 macrophages, αKG suppressed the NF-κB pathway by inhibiting IKK through PHD-dependent proline hydroxylation, which led to impaired proinflammatory responses [28][44]. Glutaminolysis is not only implicated in macrophage polarization, but also plays a role in the differentiation and function of immunosuppressive myeloid cells. Both Wu et al. [29][45] and Sun et al. [30][46] demonstrated the presence of hematopoietic progenitor cells (HPC) in cancers that give rise to immature myeloid cells (IMC) and MDSCs.
3.2. Tryptophan
It is well established that the tryptophan catabolic activity of IDO in TAMs impairs T cell activation
[31][48]. Su et al. have demonstrated that the induction of the IDO expression in TAMs is triggered by the sensing of phagocytosed tumor DNA by the AIM2 inflammasome, linking antibody-dependent cell phagocytosis (ADCP) in macrophages to the suppression of CD8 T cells and to antibody-dependent cell cytotoxicity (ADCC) in natural killer (NK) cells
[32][49]. AIM2-dependent IL-1
β does not only upregulate IDO but also the PD-L1 expression leading to immunosuppression, as shown in a breast cancer mouse model. Consistently, the inhibition of IDO in combination with anti-PD-L1 potentiated the therapeutic efficacy of the anti-HER2 antibody, completely reversing the suppressive effects of ADCP and the enhanced anti-tumor immunity
[32][49]. Kyn, the product of the IDO-derived tryptophan metabolism, has been identified as a ligand of the transcription factor aryl hydrocarbon receptor (AHR)
[33][50] (
Figure 2). In glioblastoma, Kyn produced by glioma cells activates AHR in TAMs leading to enhanced recruitment via CCR2 upregulation
[34][51]. AHR promotes macrophage immunosuppressive functions through several mechanisms, including KLF4 upregulation of M2 gene expression, repression of NF-κB via SOCS2-TRAF6 signaling, and immunosuppressive adenosine production through the induction of the ectonucleotidase CD39 (
Figure 2).
3.3. Arginine
Arginine metabolism is an important determinant of macrophage phenotypes and functions. Arginine catabolism to nitric oxide (NO) and citrulline by inducible nitric oxide synthetase (iNOS) (
Figure 2) is an essential pathway for inflammatory macrophage phagocytic, cytotoxic and anti-tumoral functions. Indeed, besides favoring a pro-inflammatory state in macrophages via NO
[35][53], arginine metabolism by iNOS triggers anti-tumor immunity via citrulline. A recent report has demonstrated that citrullination enhances the immunogenicity of epitopes presented on Major Histocompatibility Complex (MHC) class II (MHC-II), leading to an improved anti-tumoral immunity against established tumors, associated with enhanced T helper (Th)1 responses, decreased infiltration of MDSCs, and a memory response upon tumor rechallenge
[36][54]. On the other hand, arginine metabolism to ornithine, a precursor of polyamines, by ARG1 (
Figure 2) is necessary for macrophage homeostatic functions and is usurped in TAMs to support tumor growth
[37][55]. Early studies have shown that TAMs expressing high levels of ARG1 are key effectors of tumor immune evasion, and that the inhibition of ARG1 reduces the tumor growth, as demonstrated in the LLC transplantable mouse model
[38][56].
3.4. Cystine, Glutamate and Oxidative Stress
Another amino acid involved in immunoregulation in the TME is cysteine. Early work has demonstrated that one of the mechanisms used by MDSCs to inhibit T cell activity relies on cysteine homeostasis. Indeed, MDSCs and macrophages, but not T cells, express the cystine/glutamate transporter xCT (SLC7A11 or system X
c-) on their surface, allowing them to import extracellular cystine, a major oxidized form of cysteine, but without releasing cysteine back to the TME (
Figure 1). Through this action, they deprive T cells of cysteine that is necessary for their activity and function
[39][59]; cystine import regulates the cellular cysteine levels of the T cell and maintains the reduced glutathione (GSH) pool that is necessary to counter oxidative stress.
In macrophages, ROS generated by succinate oxidation drives pro-inflammatory macrophage rewiring into an M1 phenotype and promotes the pro-inflammatory cytokine production
[40][60]. However, in cancer, ROS-induced inflammatory signaling in TAMs and MDSCs favors tumorigenesis and metastasis. Liang et al. have used the diethylnitrosamine (DEN) mouse model of inflammation-driven liver cancer to demonstrate a key role of ROS and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)1 expression in macrophages in promoting liver tumorigenesis (
Figure 2).
4. Lipids and Cholesterol Metabolism Shape Immunosuppressive Myeloid Cells in the TME
Several studies have now confirmed a dependency of M2 macrophages, TAMs and MDSCs on lipid and cholesterol metabolisms. Inhibiting the lipid transport, lipolysis and FAO were shown to blunt their immunosuppressive and pro-tumoral activities. Mechanistically, the transcriptional activities of STAT6 downstream of IL-4, and of the peroxisome proliferator-activated receptor Gamma (PPAR
γ) in conjunction with its co-activator PPAR
γ-coactivator-1beta (PGC-1
β), are key for the differentiation and activation of tumor-associated myeloid cells (
Figure 2). Collectively, they function by upregulating genes involved in lipid uptake, e.g., CD36, mitochondrial biogenesis, OXPHOS and anti-inflammatory effectors. Initially, Chawla and coworkers demonstrated in vitro that PGC-1
β was induced by STAT6 downstream of IL-4 signaling and that the uncoupling of mitochondrial respiration inhibited the expression of M2 genes such as ARG1. They also showed that the deletion of STAT6 or the downregulation of PGC-1
β impaired the M2 macrophage program
[41][69]. In the same system, Pearce and colleagues showed that CD36-mediated uptake of triacylglycerol substrates and their lipolysis in the lysosome via by lysosomal acid lipase (LAL) were important in driving FAO, and that the inhibition of lipases using the weight-loss agent Orlistat blunted the M2 macrophage markers expression and M2 protective responses in the
Heligmosomoides polygyrus helminth infection mouse model
[42][70]. In cancer, polyunsaturated fatty acids (PUFAs) promote the expansion and immunosuppressive function of MDSCs through ROS production and JAK-STAT3 activation
[43][71] (
Figure 2).
5. Myelosuppressive Effects of Adenosine
A characteristic of the TME is the high levels of the immunosuppressive metabolite adenosine. Adenosine accumulates due to the rapid catabolism of ATP released by myeloid cells and dying tumor cells by the ectonucleotidases CD39 and CD73. CD39 converts ATP and ADP to AMP whereas CD73 converts the latter to adenosine. Adenosine can act directly on T cells to blunt their anti-tumoral activity through binding to its receptor, the A2A adenosine receptor (A2AR)
[44][45][80,81]. The expression of A2AR on myeloid cells, including macrophages, DCs and MDSCs, also contributes to immunosuppression in the TME (
Figure 1). This has been demonstrated with myeloid-specific deletion of
A2ar, which resulted in enhanced T cells and NK cells of anti-tumoral activity and reduced melanoma metastasis in mice
[46][82]. The adenosine generated by cancer cells is capable of stimulating macrophage migration, as demonstrated in an in vitro coculture system using ovarian cancer cell lines. In vivo, macrophages upregulate CD39 and CD73 expression in the TME, and CD73 expression is also provided by stromal fibroblasts
[47][83].
6. Wnt Ligands Potentiate Myeloid Cells—Cancer Cells Metabolic Crosstalk
The Wnt pathway is an essential homeostatic pathway regulating several processes in embryogenesis and in adult tissues, notably cellular proliferation, migration and stemness, and is one of the central pathways upregulated in cancer. Besides driving tumorigenesis, the sustained activation of the Wnt pathway is also associated with a resistance to cancer therapies, as reviewed in
[48][85]. Wnt signaling is engaged when one of 19 secreted Wnt ligands bind to members of the Frizzled receptors family, leading to the stabilization of
β-catenin and downstream transcriptional responses. Macrophages have emerged as an important source of Wnt ligands, contributing to tumor growth and progression by driving the Wnt/
β-catenin activation in cancer cells, as reviewed in
[49][86]. In a feed-forward mechanism, tumor-cell-derived Wnt ligands promote the M2 polarization of macrophages. For instance, Yang et al. have shown that the depletion of Wntless from a hepatocellular carcinoma (HCC) cell line (Hepa1-6) inhibited M2-like TAMs in the TME following orthotopic transplantation, due to the impaired Wnt/
β-catenin activation in TAMs
[50][87].
78. Conclusions and Perspectives
Targeting the metabolism of immunosuppressive myeloid cells in the TME is a promising therapeutic strategy. Besides depriving myeloid cells of the essential nutrients required for their effector functions, cancer cells release a panoply of metabolites, including lactate and adenosine, to attract myeloid cells to the TME and rewire their metabolism from an anti-tumoral to a pro-tumoral one. In addition to providing trophic functions, promoting angiogenesis and inhibiting cytotoxic T cells and NK cells tumorilytic activities, TAMs and MDSCs further support the metabolism of cancer cells by releasing metabolites, cytokines or vesicles containing lncRNAs that reinforce the Warburg effect. The identification of central metabolic nodes and effectors, such as metabolite transporters, metabolic enzymes, transcription factors, etc., provide an entry point to curb such metabolic interdependence. The recent works reviewed here provide compelling evidence for the potential in targeting the cellular metabolism to boost the efficacy of immunotherapies, which hopefully will translate to the clinic soon.