 +1 credit
+1 credit
Video Upload Options
COPD, a chronic obstructive pulmonary disease, is one of the leading causes of death worldwide. Clinical studies and research in rodent models demonstrated that failure of repair mechanisms to cope with increased ROS and inflammation in the lung leads to COPD. Despite this progress, the molecular mechanisms underlying the development of COPD remain poorly understood, resulting in a lack of effective treatments. Thus, an informative, simple model is highly valued and desired. Recently, the cigarette smoke-induced Drosophila COPD model showed a complex set of pathological phenotypes that resemble those seen in human COPD patients. The Drosophila trachea has been used as a premier model to reveal the mechanisms of tube morphogenesis. The association of these mechanisms to structural changes in COPD can be analyzed by using Drosophila trachea.
1. Urgent Need to Reveal Novel Mechanisms of COPD
2. Drosophila Trachea as a Model to Reveal Underlying Mechanisms of Tube Morphogenesis
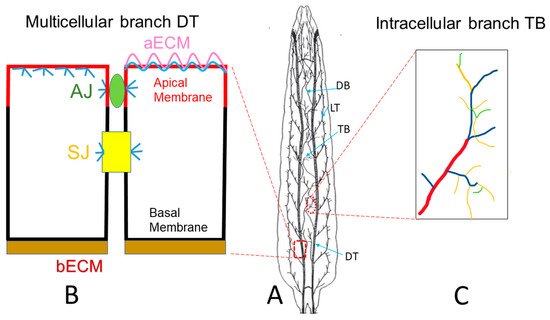
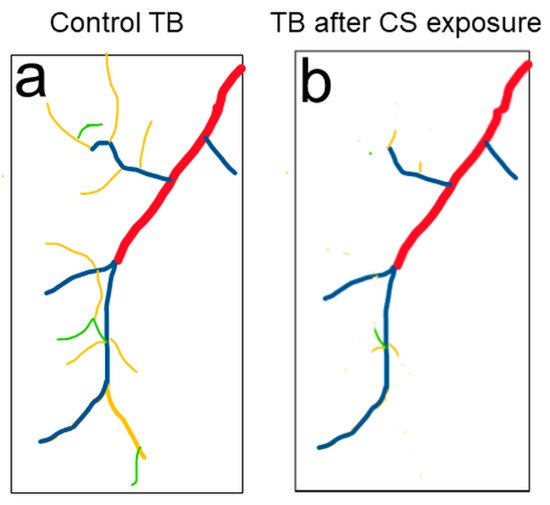
3. Drosophila Trachea as a COPD Model to Systematically Study the Development of COPD
References
- Viegi, G.; Maio, S.; Fasola, S.; Baldacci, S. Global Burden of Chronic Respiratory Diseases. J. Aerosol Med. Pulm. Drug Deliv. 2020, 33, 171–177.
- Terzikhan, N.; Verhamme, K.M.; Hofman, A.; Stricker, B.H.; Brusselle, G.G.; Lahousse, L. Prevalence and incidence of COPD in smokers and non-smokers: The Rotterdam Study. Eur. J. Epidemiol. 2016, 31, 785–792.
- Liu, S.; Jørgensen, J.T.; Ljungman, P.; Pershagen, G.; Bellander, T.; Leander, K.; Magnusson, P.K.E.; Rizzuto, D.; Hvidtfeldt, U.A.; Raaschou-Nielsen, O.; et al. Long-term exposure to low-level air pollution and incidence of chronic obstructive pulmonary disease: The ELAPSE project. Environ. Int. 2021, 146, 106267.
- Hendryx, M.; Luo, J.; Chojenta, C.; Byles, J.E. Air pollution exposures from multiple point sources and risk of incident chronic obstructive pulmonary disease (COPD) and asthma. Environ. Res. 2019, 179, 108783.
- Wiegman, C.H.; Li, F.; Ryffel, B.; Togbe, D.; Chung, K.F. Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11, 1957.
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755.
- López-Campos, J.L.; Tan, W.; Soriano, J.B. Global burden of COPD. Respirology 2016, 21, 14–23.
- Kluchova, Z.; Petrasova, D.; Joppa, P.; Dorkova, Z.; Tkacova, R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol. Res. 2007, 56, 51–56.
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27.
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, 205.
- Baarsma, H.A.; Skronska-Wasek, W.; Mutze, K.; Ciolek, F.; Wagner, D.E.; John-Schuster, G.; Heinzelmann, K.; Gunther, A.; Bracke, K.R.; Dagouassat, M.; et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J. Exp. Med. 2017, 214, 143–163.
- Barnes, P.J. The cytokine network in asthma and chronic obstructive pulmonary disease. J. Clin. Investig. 2008, 118, 3546–3556.
- Barnes, P.J. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2018, 18, 454–466.
- Tuder, R.M.; McGrath, S.; Neptune, E. The pathobiological mechanisms of emphysema models: What do they have in common? Pulm. Pharmacol. Ther. 2003, 16, 67–78.
- Martinez, F.J.; Donohue, J.F.; Rennard, S.I. The future of chronic obstructive pulmonary disease treatment--difficulties of and barriers to drug development. Lancet 2011, 378, 1027–1037.
- Michaudel, C.; Mackowiak, C.; Maillet, I.; Fauconnier, L.; Akdis, C.A.; Sokolowska, M.; Dreher, A.; Tan, H.T.; Quesniaux, V.F.; Ryffel, B.; et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J. Allergy Clin. Immunol. 2018, 142, 942–958.
- Nishida, K.; Brune, K.A.; Putcha, N.; Mandke, P.; O’Neal, W.K.; Shade, D.; Srivastava, V.; Wang, M.; Lam, H.; An, S.S.; et al. Cigarette smoke disrupts monolayer integrity by altering epithelial cell-cell adhesion and cortical tension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L581–L591.
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169.
- Wangler, M.F.; Yamamoto, S.; Chao, H.T.; Posey, J.E.; Westerfield, M.; Postlethwait, J.; Members of the Undiagnosed Diseases Network, (UDN); Hieter, P.; Boycott, K.M.; Campeau, P.M.; et al. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics 2017, 207, 9–27.
- Schneider, D. Using Drosophila as a model insect. Nat. Rev. Genet. 2000, 1, 218–226.
- Ghabrial, A.; Luschnig, S.; Metzstein, M.M.; Krasnow, M.A. Branching morphogenesis of the Drosophila tracheal system. Annu. Rev. Cell Dev. Biol. 2003, 19, 623–647.
- Ohshiro, T.; Emori, Y.; Saigo, K. Ligand-dependent activation of breathless FGF receptor gene in Drosophila developing trachea. Mech. Dev. 2002, 114, 3–11.
- Zuo, L.; Iordanou, E.; Chandran, R.R.; Jiang, L. Novel mechanisms of tube-size regulation revealed by the Drosophila trachea. Cell Tissue Res. 2013, 354, 343–354.
- Ruhle, H. Das larvale Tracheensystem von Drosophila melanogaster Meigen und seine Variabilita. Z. Wiss. Zool. 1932, 159–245.
- Devine, W.P.; Lubarsky, B.; Shaw, K.; Luschnig, S.; Messina, L.; Krasnow, M.A. Requirement for chitin biosynthesis in epithelial tube morphogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 17014–17019.
- Tonning, A.; Hemphala, J.; Tang, E.; Nannmark, U.; Samakovlis, C.; Uv, A. A transient luminal chitinous matrix is required to model epithelial tube diameter in the Drosophila trachea. Dev. Cell 2005, 9, 423–430.
- Behr, M.; Wingen, C.; Wolf, C.; Schuh, R.; Hoch, M. Wurst is essential for airway clearance and respiratory-tube size control. Nat. Cell Biol. 2007, 9, 847–853.
- Tsarouhas, V.; Senti, K.A.; Jayaram, S.A.; Tiklova, K.; Hemphala, J.; Adler, J.; Samakovlis, C. Sequential pulses of apical epithelial secretion and endocytosis drive airway maturation in Drosophila. Dev. Cell 2007, 13, 214–225.
- Petkau, G.; Wingen, C.; Jussen, L.C.; Radtke, T.; Behr, M. Obstructor-A is required for epithelial extracellular matrix dynamics, exoskeleton function, and tubulogenesis. J. Biol. Chem. 2012, 287, 21396–21405.
- Dong, B.; Hayashi, S. Shaping of biological tubes by mechanical interaction of cell and extracellular matrix. Curr. Opin. Genet. Dev. 2015, 32, 129–134.
- Armbruster, K.; Luschnig, S. The Drosophila Sec7 domain guanine nucleotide exchange factor protein Gartenzwerg localizes at the cis-Golgi and is essential for epithelial tube expansion. J. Cell Sci. 2012, 125, 1318–1328.
- Wang, S.; Meyer, H.; Ochoa-Espinosa, A.; Buchwald, U.; Onel, S.; Altenhein, B.; Heinisch, J.J.; Affolter, M.; Paululat, A. GBF1 (Gartenzwerg)-dependent secretion is required for Drosophila tubulogenesis. J. Cell Sci. 2012, 125, 461–472.
- Llimargas, M.; Strigini, M.; Katidou, M.; Karagogeos, D.; Casanova, J. Lachesin is a component of a septate junction-based mechanism that controls tube size and epithelial integrity in the Drosophila tracheal system. Development 2004, 131, 181–190.
- Wu, V.M.; Schulte, J.; Hirschi, A.; Tepass, U.; Beitel, G.J. Sinuous is a Drosophila claudin required for septate junction organization and epithelial tube size control. J. Cell Biol. 2004, 164, 313–323.
- Wilkin, M.B.; Becker, M.N.; Mulvey, D.; Phan, I.; Chao, A.; Cooper, K.; Chung, H.J.; Campbell, I.D.; Baron, M.; MacIntyre, R. Drosophila dumpy is a gigantic extracellular protein required to maintain tension at epidermal-cuticle attachment sites. Curr. Biol. 2000, 10, 559–567.
- Zhang, L.; Ward, R.E. uninflatable encodes a novel ectodermal apical surface protein required for tracheal inflation in Drosophila. Dev. Biol. 2009, 336, 201–212.
- Hannezo, E.; Dong, B.; Recho, P.; Joanny, J.F.; Hayashi, S. Cortical instability drives periodic supracellular actin pattern formation in epithelial tubes. Proc. Natl. Acad. Sci. USA 2015, 112, 8620–8625.
- Matusek, T.; Djiane, A.; Jankovics, F.; Brunner, D.; Mlodzik, M.; Mihaly, J. The Drosophila formin DAAM regulates the tracheal cuticle pattern through organizing the actin cytoskeleton. Development 2006, 133, 957–966.
- Araujo, S.J.; Cela, C.; Llimargas, M. Tramtrack regulates different morphogenetic events during Drosophila tracheal development. Development 2007, 134, 3665–3676.
- Hemphala, J.; Uv, A.; Cantera, R.; Bray, S.; Samakovlis, C. Grainy head controls apical membrane growth and tube elongation in response to Branchless/FGF signalling. Development 2003, 130, 249–258.
- Gervais, L.; Casanova, J. In vivo coupling of cell elongation and lumen formation in a single cell. Curr. Biol. 2010, 20, 359–366.
- Best, B.T. Single-cell branching morphogenesis in the Drosophila trachea. Dev. Biol. 2019, 451, 5–15.
- Jones, T.A.; Metzstein, M.M. A novel function for the PAR complex in subcellular morphogenesis of tracheal terminal cells in Drosophila melanogaster. Genetics 2011, 189, 153–164.
- Ozturk-Colak, A.; Moussian, B.; Araujo, S.J. Drosophila chitinous aECM and its cellular interactions during tracheal development. Dev. Dyn. 2016, 245, 259–267.
- Jones, T.A.; Nikolova, L.S.; Schjelderup, A.; Metzstein, M.M. Exocyst-mediated membrane trafficking is required for branch outgrowth in Drosophila tracheal terminal cells. Dev. Biol. 2014, 390, 41–50.
- Mathew, R.; Rios-Barrera, L.D.; Machado, P.; Schwab, Y.; Leptin, M. Transcytosis via the late endocytic pathway as a cell morphogenetic mechanism. EMBO J. 2020, 39, e105332.
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13.
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162.
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12.
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611.
- Fu, X.; Zhang, F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp. Ther. Med. 2018, 16, 4553–4561.
- Bacon, N.C.; Wappner, P.; O’Rourke, J.F.; Bartlett, S.M.; Shilo, B.; Pugh, C.W.; Ratcliffe, P.J. Regulation of the Drosophila bHLH-PAS protein Sima by hypoxia: Functional evidence for homology with mammalian HIF-1 alpha. Biochem. Biophys. Res. Commun. 1998, 249, 811–816.
- Centanin, L.; Dekanty, A.; Romero, N.; Irisarri, M.; Gorr, T.A.; Wappner, P. Cell Autonomy of HIF Effects in Drosophila: Tracheal Cells Sense Hypoxia and Induce Terminal Branch Sprouting. Dev. Cell 2008, 14, 547–558.
- Habib, P.; Jung, J.; Wilms, G.M.; Kokott-Vuong, A.; Habib, S.; Schulz, J.B.; Voigt, A. Posthypoxic behavioral impairment and mortality of Drosophila melanogaster are associated with high temperatures, enhanced predeath activity and oxidative stress. Exp. Mol. Med. 2021, 53, 264–280.
- Sato, M.; Kornberg, T.B. FGF is an essential mitogen and chemoattractant for the air sacs of the drosophila tracheal system. Dev. Cell 2002, 3, 195–207.
- Cruz, J.; Bota-Rabassedas, N.; Franch-Marro, X. FGF coordinates air sac development by activation of the EGF ligand Vein through the transcription factor PntP2. Sci. Rep. 2015, 5, 17806.
- Cabernard, C.; Affolter, M. Distinct Roles for Two Receptor Tyrosine Kinases in Epithelial Branching Morphogenesis in Drosophila. Dev. Cell 2005, 9, 831–842.
- Chanut-Delalande, H.; Jung, A.C.; Baer, M.M.; Lin, L.; Payre, F.; Affolter, M. The Hrs/Stam Complex Acts as a Positive and Negative Regulator of RTK Signaling during Drosophila Development. PLoS ONE 2010, 5, e10245.
- Dong, Q.; Brenneman, B.; Fields, C.; Srivastava, A. A Cathepsin-L is required for invasive behavior during Air Sac Primordium development inDrosophila melanogaster. FEBS Lett. 2015, 589, 3090–3097.
- Llano, E.; Adam, G.; Pendás, A.M.; Quesada, V.; Sánchez, L.M.; Santamaría, I.; Noselli, S.; López-Otín, C. Structural and Enzymatic Characterization of Drosophila Dm2-MMP, a Membrane-bound Matrix Metalloproteinase with Tissue-specific Expression. J. Biol. Chem. 2002, 277, 23321–23329.
- Guha, A.; Lin, L.; Kornberg, T.B. Regulation of Drosophila matrix metalloprotease Mmp2 is essential for wing imaginal disc:trachea association and air sac tubulogenesis. Dev. Biol. 2009, 335, 317–326.
- Park, W.Y.; Miranda, B.; Lebeche, D.; Hashimoto, G.; Cardoso, W.V. FGF-10 Is a Chemotactic Factor for Distal Epithelial Buds during Lung Development. Dev. Biol. 1998, 201, 125–134.
- Abler, L.L.; Mansour, S.L.; Sun, X. Conditional gene inactivation reveals roles forFgf10andFgfr2in establishing a normal pattern of epithelial branching in the mouse lung. Dev. Dyn. 2009, 238, 1999–2013.
- Brandsma, C.A.; de Vries, M.; Costa, R.; Woldhuis, R.R.; Konigshoff, M.; Timens, W. Lung ageing and COPD: Is there a role for ageing in abnormal tissue repair? Eur. Respir. Rev. 2017, 26, 170073.
- McDonough, J.E.; Yuan, R.; Suzuki, M.; Seyednejad, N.; Elliott, W.M.; Sanchez, P.G.; Wright, A.C.; Gefter, W.B.; Litzky, L.; Coxson, H.O.; et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N. Engl. J. Med. 2011, 365, 1567–1575.
- Yew-Booth, L.; Birrell, M.A.; Lau, M.S.; Baker, K.; Jones, V.; Kilty, I.; Belvisi, M.G. JAK-STAT pathway activation in COPD. Eur. Respir. J. 2015, 46, 843–845.
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2008, 39, 673–682.
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474.
- Vogelmeier, C.F.; Roman-Rodriguez, M.; Singh, D.; Han, M.K.; Rodriguez-Roisin, R.; Ferguson, G.T. Goals of COPD treatment: Focus on symptoms and exacerbations. Respir. Med. 2020, 166, 105938.
- Santalla, M.; Pagola, L.; Gómez, I.; Balcazar, D.; Valverde, C.A.; Ferrero, P. Smoking flies: Testing the effect of tobacco cigarettes on heart function of Drosophila melanogaster. Biol. Open 2021, 10, bio055004.
- Giannopoulos, A.; Giannakou, L.; Gourgouliani, N.; Lüpold, S.; Rouka, E.; Jagirdar, R.; Pitaraki, E.; Hatzoglou, C.; Gourgoulianis, K.; Blanckenhorn, W.; et al. Exposure of Drosophila melanogaster to cigarette smoke extract changes its sexual behavior. Eur. Respir. J. 2020, 56, 1326.
- Morris, M.; Shaw, A.; Lambert, M.; Perry, H.H.; Lowenstein, E.; Valenzuela, D.; Velazquez-Ulloa, N.A. Developmental nicotine exposure affects larval brain size and the adult dopaminergic system of Drosophila melanogaster. BMC Dev. Biol. 2018, 18, 13–16.
- Prange, R.; Thiedmann, M.; Bhandari, A.; Mishra, N.; Sinha, A.; Hasler, R.; Rosenstiel, P.; Uliczka, K.; Wagner, C.; Yildirim, A.O.; et al. A Drosophila model of cigarette smoke induced COPD identifies Nrf2 signaling as an expedient target for intervention. Aging 2018, 10, 2122–2135.
- Pfeiffenberger, C.; Lear, B.C.; Keegan, K.P.; Allada, R. Locomotor activity level monitoring using the Drosophila Activity Monitoring (DAM) System. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5518.
- Hoffmann, J.; Romey, R.; Fink, C.; Yong, L.; Roeder, T. Overexpression of Sir2 in the adult fat body is sufficient to extend lifespan of male and female Drosophila. Aging 2013, 5, 315–327.
- Wu, V.M.; Yu, M.H.; Paik, R.; Banerjee, S.; Liang, Z.; Paul, S.M.; Bhat, M.A.; Beitel, G.J. Drosophila Varicose, a member of a new subgroup of basolateral MAGUKs, is required for septate junctions and tracheal morphogenesis. Development 2007, 134, 999–1009.
- Kiss, M.; Kiss, A.A.; Radics, M.; Popovics, N.; Hermesz, E.; Csiszar, K.; Mink, M. Drosophila type IV collagen mutation associates with immune system activation and intestinal dysfunction. Matrix Biol. 2016, 49, 120–131.
- Ling, C.; Zheng, Y.; Yin, F.; Yu, J.; Huang, J.; Hong, Y.; Wu, S.; Pan, D. The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc. Natl. Acad. Sci. USA 2010, 107, 10532–10537.
- Khanna, M.R.; Mattie, F.J.; Browder, K.C.; Radyk, M.D.; Crilly, S.E.; Bakerink, K.J.; Harper, S.L.; Speicher, D.W.; Thomas, G.H. Spectrin tetramer formation is not required for viable development in Drosophila. J. Biol. Chem. 2015, 290, 706–715.
- Kessenbrock, K.; Holak, T.A.; Sixt, M.; Jenne, D.; Werb, Z.; Riedl, J.; Wedlich-Soldner, R.; Neukirchen, D.; Bista, M.; Crevenna, A.H.; et al. Lifeact: A versatile marker to visualize F-actin. Nat. Methods 2008, 5, 605–607.
- Chen, Y.J.; Huang, J.; Huang, L.; Austin, E.; Hong, Y. Phosphorylation potential of Drosophila E-Cadherin intracellular domain is essential for development and adherens junction biosynthetic dynamics regulation. Development 2017, 144, 1242–1248.
- Ghabrial, A.S.; Krasnow, M.A. Social interactions among epithelial cells during tracheal branching morphogenesis. Nature 2006, 441, 746–749.
- Liu, Y.; Wei, H.; Tang, J.; Yuan, J.; Wu, M.; Yao, C.; Hosoi, K.; Yu, S.; Zhao, X.; Han, Y.; et al. Dysfunction of pulmonary epithelial tight junction induced by silicon dioxide nanoparticles via the ROS/ERK pathway and protein degradation. Chemosphere 2020, 255, 126954.
- Feng, S.; Zou, L.; Wang, H.; He, R.; Liu, K.; Zhu, H. RhoA/ROCK-2 Pathway Inhibition and Tight Junction Protein Upregulation by Catalpol Suppresses Lipopolysaccaride-Induced Disruption of Blood-Brain Barrier Permeability. Molecules 2018, 23, 2371.
- Landis, G.; Shen, J.; Tower, J. Gene expression changes in response to aging compared to heat stress, oxidative stress and ionizing radiation in Drosophila melanogaster. Aging 2012, 4, 768–789.
- Dar, N.J.; Satti, N.K.; Dutt, P.; Hamid, A.; Ahmad, M. Attenuation of Glutamate-Induced Excitotoxicity by Withanolide-A in Neuron-Like Cells: Role for PI3K/Akt/MAPK Signaling Pathway. Mol. Neurobiol. 2018, 55, 2725–2739.
- Yao, Y.; Zhou, J.; Diao, X.; Wang, S. Association between tumor necrosis factor-alpha and chronic obstructive pulmonary disease: A systematic review and meta-analysis. Ther. Adv. Respir. Dis. 2019, 13, 1753466619866096.
- Moreno, E.; Yan, M.; Basler, K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 2002, 12, 1263–1268.
- Igaki, T.; Kanda, H.; Yamamoto-Goto, Y.; Kanuka, H.; Kuranaga, E.; Aigaki, T.; Miura, M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002, 21, 3009–3018.
- Boulay, J.L.; O’Shea, J.J.; Paul, W.E. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 2003, 19, 159–163.
- Brown, S.; Hu, N.; Hombria, J.C. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr. Biol. 2001, 11, 1700–1705.
- Chen, H.W.; Chen, X.; Oh, S.W.; Marinissen, M.J.; Gutkind, J.S.; Hou, S.X. mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes Dev. 2002, 16, 388–398.
- Çolak, Y.; Afzal, S.; Nordestgaard, B.G.; Lange, P.; Vestbo, J. Importance of Early COPD in Young Adults for Development of Clinical COPD: Findings from the Copenhagen General Population Study. Am. J. Respir. Crit. Care Med. 2021, 203, 1245–1256.

