 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Saverio Bellusci | + 5370 word(s) | 5370 | 2020-08-13 10:10:16 | | | |
| 2 | Conner Chen | -171 word(s) | 5199 | 2020-08-20 10:46:24 | | | | |
| 3 | Conner Chen | -1 word(s) | 5198 | 2020-08-26 08:37:27 | | |
Video Upload Options
Bronchopulmonary dysplasia (BPD) is a chronic lung disease affecting preterm infants. More than 50 years after the first description of BPD by Northway, this chronic lung disease affecting many preterm infants is still less understood. Additonally, approximately 40% of preterm infants suffering from severe BPD also suffer from Bronchopulmonary dysplasia-associated pulmonary hypertension (BPD-PH), leading to a significant increase in total morbidity and mortality. Until today, there is no curative therapy for both BPD and BPD-PH available. It has become increasingly evident that growth factors are playing a central role in normal and pathologic development of the pulmonary vasculature. Thus, this review aims to summarize the recent evidence in our understanding of BPD-PH from a basic scientific point of view, focusing on the potential role of Fibroblast Growth Factor (FGF)/FGF10 signaling pathway contributing to disease development, progression and resolution.
1. Introduction
There is a rising incidence of preterm births which is one of the leading cause of death in infants younger than 5 years of age [1][2]. Bronchopulmonary dysplasia (BPD) is a common complication for infants born before 30 weeks of gestational age. The incidence of BPD is approximately 40%. BPD contributes substantially to long-term morbidities and mortalities [3]. Currently, there is no curative therapy for BPD.
BPD is a complex chronic lung disease caused by the interplay of lung development and pre- and postnatal injurious events such as ventilatory damages, oxygen toxicity, infectious stimuli, growth restriction, and repair/remodeling processes. Due to advances in the clinical management of preterm infants (e.g., use of exogenous surfactant, antenatal administration of steroids, more gentle non-invasive ventilation, and nutritional strategies in the clinical management of preterm infants) the mortality has reduced but the proportion of very early preterm infants has increased. During the so-called post-surfactant era, remarkable changes regarding the histomorphology of fatal BPD have been observed. The ”new“ BPD is characterized by simplification of alveolar formation and arrested microvascular development as compared with fibroproliferative processes mostly of the airways in ”old“ BPD. Whether these histomorphological changes result from different windows of injury (very early stages versus late stages of lung development) or from different treatment approaches (e.g., harsh mechanical ventilation with high oxygen versus non-invasive ventilation) is still unclear.
Histomorphologically, BPD is mainly characterized as alveolar simplification and pulmonary vascular remodeling. Pre- and postnatal pulmonary inflammatory responses (e.g., chorioamnionitis) are central risk factors for developing BPD. Pulmonary inflammatory responses can be induced by oxygen therapy or mechanical ventilation leading to the imbalance of proinflammatory and anti-inflammatory cytokines. This is associated with an accumulation of inflammatory cells in the lung. For example, it has been demonstrated that interleukin 10 (IL-10) and growth factors such as vascular endothelial growth factor alpha (VEGFA), platelet derived growth factor alpha (PDGFA), and fibroblast growth factors (FGFs) are decreased in tracheal aspirates and lung tissue [4][5][6].
Pulmonary hypertension (PH) is a common and life-threatening complication in BPD. A recent meta-analysis including 1400 preterm infants revealed that the prevalence of PH increases correspondingly with the severity of BPD. The prevalence is estimated to be approximately 40% in preterm infants suffering from severe BPD [7][8].
Pulmonary vascular remodeling (VR) underlying PH leads to (I) increased muscularization of already muscularized proximal and middle-sized vessels due to proliferation of existing vascular smooth muscle cells (VSMCs) and (II) de novo muscularization of normally non-muscularized distal vessels. The latter are typically the distal arterioles with diameters under 30 µm. VR results in increased pressure in the pulmonary circulation leading to an increased right ventricle (RV) afterload and potentially fatal RV hypertrophy (RVH) [9][10][11]. It needs to be mentioned that the cellular origin of the newly formed VSMCs in BPD is still elusive. The complications associated with BPD without PH includes a higher risk for developing susceptibility to infections of the upper and lower respiratory tract and asthma. By contrast, infants suffering from BPD-PH are of higher risk for oxygen dependency and right ventricle hypertrophy, which requires long-term medication and follow-up. In the worst case, these additional complications can lead to right heart failure. The lifelong morbidity of patients suffering from BPD-PH represents a substantial and growing burden for patients and health care systems [12][13].
Although recent guidelines published by the American Heart Association (AHA), American Thoracic Society (ATS), the European Pediatric Pulmonary Vascular Disease Network (PVD), and the Pediatric Pulmonary Hypertension Network (PPHNet) encourage and improve the implementation of standardized diagnostic and treatment protocols, the molecular and cellular pathomechanisms underlying BPD-associated PH remain poorly characterized, reflected by the rather limited pharmacotherapeutic options available to mitigate but not cure BPD-PH [14][15][16]
2. Development of Normal Pulmonary Vasculature and BPD-PH from the Basic Scientific Point-of-View
According to the Fifth World Symposium held in France, PH is clinically classified into five groups (Group 1, pulmonary arterial hypertension (PAH); Group 2, pulmonary hypertension due to left heart disease; Group 3, pulmonary hypertension due to chronic lung disease or hypoxia; Group 4, chronic thromboembolic pulmonary hypertension; and Group 5, pulmonary hypertension due to unclear multifactorial mechanisms) [17][18]. Groups 1, 3 and 4 are all defined as precapillary pulmonary hypertension [19]. Therefore, they share similar molecular and cellular mechanisms and histopathology. Pediatric PH has some common features with adult PH but also shows its own features. BPD-PH, characterized by impaired alveolar growth and distorted pulmonary vascular development, is categorized as a subgroup of Group 3 (Group 3.5, developmental lung disorders) [20]. Therefore, in the next section, we describe the different lung vascular growth abnormalities and the potential reasons of BPD-PH from the developmental angle.
2.1. Normal Development of Pulmonary Vasculature
2.1.1. Lung Vasculature Formation and Maturation
Human lung development can be histologically divided into four distinguishable stages: embryonic and pseudoglandular stages (human, week 4–17 and mouse, E9.5–E16.5), canalicular stage (human, week 17–26 and mouse, E16.5–E17.5), saccular stage (human, week 26–36 and mouse, E17.5–PN5), and alveolar stage (human, week 36–8 years and mouse, PN5–PN30) [21][22].
The vasculature starts to develop as early as the lung buds evaginate from the foregut endoderm [23]. At E10, angioblasts and hematopoietic cells, localized at the distal mouse lung buds, form the blood islands through a processed termed vasculogenesis. The blood islands are comprised of circumferential layers of flattened angioblasts which represent primitive endothelial cells (ECs) and inner hematopoietic cells [24]. From E11 to E12, proximal vessels sprout from the main pulmonary trunk (proximal angiogenesis), run along the main conducting airways, as well as its many ramified branches. In addition, the distal blood islands increase markedly and are connected together to form the primitive capillary plexus (distal angiogenesis) [21]. During the late pseudoglandular stage, the capillary network, around lung buds, fuses with the proximal vessels. This capillary network expands significantly during the canalicular and saccular stages and gradually gets close to each other to become a double capillary layer between the adjacent lateral walls of distal sacs which form during the process of primary septa formation. During alveologenesis, concomitant with the formation of the secondary septa, the capillary network folds up to form new double capillary layers. These double capillary layers further evolve into a single capillary layer through a process called microvascular maturation. This process reduces the distance between the alveolar walls and the capillaries, thereby facilitating efficient gas exchange [25][26]. Microvascular maturation occurs concomitantly to alveologenesis and, in humans, lasts until young adulthood [27].
2.1.2. Pulmonary Vascular Development Needs Multicellular Crosstalks
Interactions among different cell types are indispensable for the formation and maturation of the vascular system during lung development. At the early stage of lung development, mesenchymal cells express Vegf, which interacts with its receptors, Vegfr1 (Flt-1), Vegfr2 (Flk-1), and Vegfr3 on endothelial cells (ECs), initiating and regulating the formation of vasculogenesis and angiogenesis [28][29]. Vegfr2 is the most important signaling receptor, which upon activation, initiates and promotes vasculogenesis and angiogenesis [30]. In contrast, Vegfr1 functions as a ligand trap, therefore, reducing the interaction between Vegf and Vegfr2, thereby decreasing the overexpansion of ECs [31]. From the late pseudoglandular stage to the canalicular stage, the cellular source of Vegf gradually shifts from the resident mesenchymal cells to the alveolar epithelial cells (mainly alveolar type 2 (AT2) cells). This results in further attraction of the capillaries towards the epithelium, thereby promoting the formation of primitive alveolar septa through EC-derived angiocrine factors, such as hepatocyte growth factor (Hgf) [32][33][34].
Following the interaction between epithelium and mesenchyme, a population of mesenchymal cells differentiate into α-smooth muscle actin (Sma)-positive mural cells (VSMCs and pericytes), another component of the blood vessel wall [35]. VSMCs, which display a flattened, spindle, and dense structure, are usually present in the media of large vessels such as arteries, arterioles, and veins. From large vessels to capillaries, VSMCs are gradually getting sparse and changing into round, protruded cells, termed pericytes. They always embed in the basement membrane and adhere tightly to ECs [36][37]. (Figure 1). In contrast to VSMCs, several markers including neuron-glial antigen-2 (Ng2), Cd146, α-Sma, Sm22, desmin, platelet-derived growth factor receptor-β (Pdgfr-β), aminopeptidase A and N, RGS5, and the promoter trap transgene XlacZ4 have been identified to label pericytes [38][39][40][41].
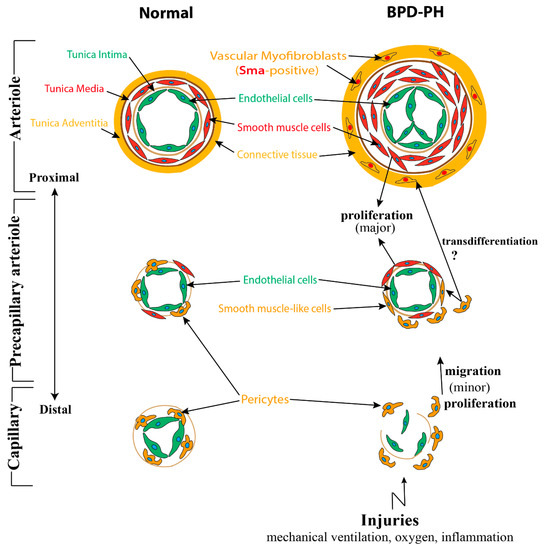
Figure 1. A speculative model of structural changes of the pulmonary vessel wall in bronchopulmonary dysplasia-associated pulmonary hypertension (BPD-PH). Under a normal situation, pulmonary arterioles are wrapped by the following three layers of tunicae: The tunica intima consists of endothelium, basement membrane, and internal elastic tissue; the tunica media is comprised of smooth muscle cells and external elastic tissue; and connective tissues constitute the tunica adventitia. From large vessels to small vessels, tunica media and adventitia are gradually getting sparse and the smooth muscle cells gradually evolving into pericytes, which adhere tightly to capillary endothelial cells (ECs). While in BPD, due to multifactorial injuries, the endothelial cells are dysfunctional and eventually become apoptosis resistant. Smooth muscle cells, which mostly proliferated from resident smooth muscle cells (SMCs) are increased significantly, resulting in the thickening of tunica media and the muscularization of normally non-muscular vessels. Adventitia of pulmonary arterioles undergo α-Sma positive myofibroblast transition assisted by altered extracellular matrix breakdown and deposition. Pericytes disconnect with capillary ECs, leading to the loss of small capillaries, proliferate, and migrate into the mesenchyme and also contribute to a small population of smooth muscle-like cells.
Pericytes play a critical role in regulating microvascular and alveolar development. Pericytes, on the one hand, adhere to capillary ECs to regulate their function, proliferation, migration, and differentiation via several signaling pathways including Vegf/Vegfr, Pdgfb/Pdgfrb2, Tgfβ/Alk, S1p/Endoglin 1(Edg-1), Ang1/2/Tie2, Cadherin, and Notch signaling pathways [42][43][44][45][46]. On the other hand, pericytes also interact with AT2 cells to promote secondary septa formation through the Hippo pathway components Yap and Taz [47]. In addition, pericytes can also function as pulmonary stem cells, upon stimulation, transdifferentiating into VSMCs and myofibroblasts [48][49].
Interactions between endothelial cells (ECs) and smooth muscle cells (SMCs) play a major role in pulmonary vasculature. Normal interplay among these two cell types control the homeostasis of the pulmonary circulation, whereas aberrant association can contribute to the pulmonary vascular diseases and pulmonary hypertension. It has been well established that the release of various vasoactive agents such as nitric oxide (NO) and endothelin-1 (Et−1) through paracrine signaling endothelial cells regulates smooth muscle cell activity. However, other non-paracrine signaling mediated crosstalk between EC and SMC (such as communication through myoendothelial junctions, as well as interaction via extracellular vesicles) exists, which is altered under the pathological condition, thus, causing an increase in vasocontractility and abnormal vascular proliferation, therefore, leading to vascular remodeling, right ventricular hypertrophy, and pulmonary hypertension [50].
2.2. Pathologic Development of Pulmonary Vasculature Leading to PH in BPD
2.2.1. Abnormalities of Pulmonary Vasculature Observed in BPD-PH
Impaired pulmonary vascular development, VR and PH are likely associated with interruption of vascular formation during lung development. However, not all premature infants develop BPD or BPD-PH. It has been claimed that genetic components (e.g., polymorphisms in matrix metallopeptidase 16 (MMP16) and SPARC (Osteonectin) Cwcv and Kazal like domains proteoglycan 2 (SPOCK2) could play a role regarding the susceptibility for BPD [51][52].
We previously found that conditional deletion of phosphatase and tensin homologue (Pten) in early embryonic mouse lung mesenchyme led to lethality at birth with disorganized alveolar capillary beds [53]. This phenotype is similar to the lethal alveolar capillary dysplasia phenotype observed in newborn babies. In support of developmental perturbations, BPD often occurs in preterm infants born before the alveolar stage, and thus susceptible to various injuries such as mechanical ventilation, hyperoxia, and airway inflammation. These, in turn, affect pulmonary vascular development, leading to vascular remodeling, PH, and impaired alveologenesis .
Bhatt et al. analyzed lung samples from infants who died with BPD versus infants who died from non-pulmonary causes and found a decreased expression of VEGF and platelet endothelial cell adhesion molecule-1 (PECAM, also termed CD31, endothelial marker), as well as a decreased staining density of alveolar capillaries in BPD infants, indicating that the development of the pulmonary microvasculature was disrupted in BPD patients. Consistent with previous results, we and others, utilizing BPD animal models, established by hyperoxia exposure, also showed a decrease of endothelial cells in capillaries and blood vessel numbers and an increase of α-Sma positive cells (VSMC) in the tunica media of pulmonary arterioles and normally non-muscularized precapillary arterioles [54][55][56]. A lineage tracing study indicated that the expansion of resident SMCs was the major source related to the thickening of the smooth muscle layer in adult PH [57]. An increased collagen and elastin expression associated with increased α-SMA-positive myofibroblasts, possibly due to endothelial to mesenchymal transition (EndMT), has also been proposed as a mechanism for the accumulation of myofibroblats in the adventitia of pulmonary arteries (Figure 2) [58][59].
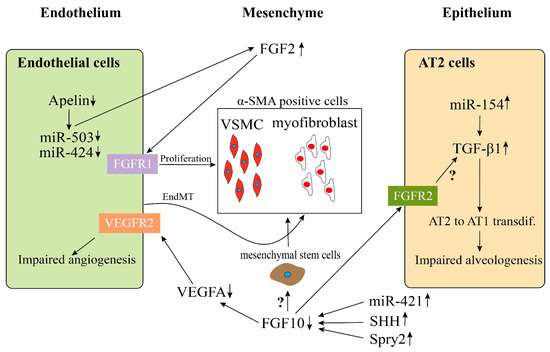
Figure 2. Possible endothelial–mesenchymal and epithelial–mesenchymal interactions in BPD-PH. Disrupted endothelial Apelin, miR-503, and miR-424 results in an increased expression of FGF2 and induces the hyperproliferation of vessel smooth muscle cells (VSMC). An increase of α-SMA-positive myofibroblasts could be due to endothelial-mesenchymal transition (EndMT). The continuous high expression of miR-154 in AT2 cells stimulate the activation of TGFβ1 signaling pathway, which results in an impaired alveologenesis. Decreased FGF10 expression in BPD, which is caused by the upregulation of miR-421, SHH, and Spry2, leads to the downregulation of VEGFA and upregulation of α-SMA, ultimately resulting in an impaired angiogenesis and an increase of α-SMA positive cells. However, whether FGF10 acts directly on the mesenchymal stem cells (MSC) to induce them to differentiate into α-SMA positive cells or through activating TGFβ1 signaling pathway needs to be further investigated.
In addition, it has also been proposed that the pericytes play a potential role in the microvascular remodeling of PH. In a clinical study, Assaad et al. found that the numbers of pericytes associated with upregulated PDGF-B expression were significantly increased in pulmonary capillary hemangiomatosis (PCH) patients, a cause of PH [60]. Furthermore, Ricard et al. also showed an increased migration and proliferation of pericytes induced by FGF2 and IL-2, in human PH patients. Finally, increased TGFβ also promotes the transdifferentiation of pericytes into contractile α-SMA positive cells (Figure 1) [61].
2.2.2. Impaired Multicellular Interactions Disrupt Pulmonary Vascular Development in BPD-PH
Multiple studies aiming at elucidating the pathogenesis of pulmonary VR have demonstrated an impairment of multicellular interactions, which could have resulted from abnormal expression levels of signaling pathways involved in cell–cell interactions.
As aforementioned, the Vegf signaling pathway is one of the most important signaling pathways regulating epithelial-endothelial crosstalk during pulmonary vascular development. Multiple studies have demonstrated that Vegf and Vegfr2 expressions were significantly decreased, while the expression of soluble Fms-like tyrosine kinase 1 (sFlt-1), an endogenous antagonist to Vegf corresponding to a truncated form of the Vegf receptor acting as a dominant negative Vegf receptor, was significantly increased, in experimental BPD animal models, which lead to a reduction and disarrangement of the microvasculature [62][63].
Other signaling pathways potentially involved in the angiogenic network include sonic hedgehog (Shh) and Sprouty2 (Spry2). Sprouty2 is an inhibitor of Fgf10. Fgf10 is known to be a major regulator of epithelial branching. Inhibition of Vegfr1-mediated signaling leads to upregulation of Spry2 in the epithelium, suggesting a downregulation of Fgf10 signaling. This is an essential evidence demonstrating the importance of endothelial-mesenchymal crosstalk. More evidence confirming the endothelial-epithelial crosstalk came from DeLisser and colleagues. Pecam1-deficient mice revealed disrupted endothelial cell formation associated with decreased alveolar simplification [64].
Furthermore, a recent study found a reduction of WNT5 in the pulmonary microvascular endothelial cells (PMVECs) of PH patients. Through exposing Wnt5a Endothelial cKO mice with chronic hypoxia, the authors demonstrated that loss of endothelium-derived Wnt5a impaired the endothelium–pericytes interaction, resulting in significant reduction, muscularization, and decreased pericyte coverage of microvessels [48]. Another study, conducted in adult PH, revealed that PH pericytes overexpressed C-X-C motif chemokine receptor-7 (CXCR-7) and TGFβRII, and as compared with control pericytes, they were more likely to proliferate, migrate, and differentiate into smooth muscle-like cells, indicating a significant role of endothelium–pericytes interaction in the process of PH (Figure 1) [65].
Pericyte-myofibroblast transition (PMT) could represent another pathogenesis of pulmonary VR, consistent with the results conducted in kidney fibrosis [66]. Wang et al. found that lung pericytes differentiated into myofibroblast in idiopathic pulmonary fibrosis (IPF) patients through increasing NOTCH1/PDGFRβ/ROCK1 signaling pathway [67]. However, the exact role of pericytes in the pathogenesis of BPD-PH is still unclear and needs to be further explored.
2.3. MicroRNAs (miRs) May Be Associated with the Pathologic Vascular Development of BPD-PH
A growing number of evidences show that expression of numerous microRNAs (miRs), that normally regulate different signaling pathways mediating cellular crosstalk in lung development, are dysregulated in PH and BPD. Thompson and Lawrie summarized a list of miRs which had therapeutic effects on PH from hypoxia or monocrotaline (MCT)-induced PH in animal experiments . Combining the differentially expressed miRs in BPD versus control with Thompson and Lawrie’s findings in PH, we identified several miRs which were all dysregulated and had the same changes in both BPD and PH (Table 1) [66][67]. These dysregulated miRs could be involved in BPD-PH.
Table 1. MicroRNAs (miRs) potentially involved in pulmonary vascular remodeling in BPD.
| miRs Shown to be Involved | Changes in PH and BPD | Potential Roles in BPD-PH | References |
|---|---|---|---|
| miR-21 | increased | Increases PASMCs proliferation through inhibiting Bmpr2 activation | [68][69] |
| miR-29 | increased | Impairs PASMCs and ECs function through down-regulating PPARγ | [70][71][72] |
| miR-34a | increased | increases lung epithelial cell apoptosis through down-regulating ANG1-TIE2 signaling; impairs alveologenesis through increasing PDGFR alpha-expressing myofibroblasts | [73][74] |
| miR-126 | decreased | Decreases microvessel density through up-regulating SPRED-1 | [69][75] |
| miR-130 | increased | Promotes ECM remodeling, increases PASMCs proliferation and crosstalk through down-regulating PPARγ-APOE-LRP8 axis | [67][76] |
| miR-154 | increased | Impairs alveologenesis through increasing TGFβ. | [77][78][79] |
| miR-421 | increased | Impairs alveologenesis through down-regulation of FGF10 signaling | [80] |
| miR-503 | decreased | Impairs ECs function and induces PASMCs proliferation through upregulating FGF2 and FGFR1 | [67][81] |
Abbreviations: PASMCs, pulmonary artery smooth muscle cells; ECs, endothelial cells; BMPR2, bone morphogenetic protein receptor type 2; PPARγ, peroxisome proliferator-activated receptor gamma; Ang, angiopoietin-1; PDGFRalpha, platelet-derived growth factor receptor alpha; SPRED-1, sprouty-related EVH1 domain containing 1; APOE, apolipoprotein E; LRP8, LDL receptor-related protein 8; TGFβ, transforming growth factor beta; FGF, fibroblast growth factor.
Furthermore, there is evidence that miRs target FGF signaling to regulate proliferation. In the study by Chen et al., the authors showed that miR-339 inhibited the proliferation of pulmonary artery smooth muscle cells (PASMCs) by targeting FGF signals [82]. By using EdU incorporation assay, they showed that miR-339 inhibited the proliferation of PASMC. In addition, they found that miR-339 inhibited FGF2-induced proliferation of PASMCs but had no effect on PDGF-BB-induced proliferation via functional analysis. Using a bioinformatics tool (Targetscan), they found that FRS2 was a potential target of miR-399 to target FGF signaling.
In disease condition, it has also been shown that the Apelin (APLN) and FGF2 pathways in pulmonary artery endothelial cells (PAECs) are regulated by miR-424 and miR-503. In PH, this pathway is disrupted (Figure 2) [81].
Yuan et al. found that the expression of miR-421 in mice exposed to hyperoxia-induced lung injury (mouse model of BPD) was much higher than that of room air control mice. However, the expression of Fgf10 decreased significantly as compared with the control group [80]. As a result, inhibition of miR-421 reduced bronchopulmonary dysplasia in mice by upregulating Fgf10 (Figure 2).
3. Role of FGF/FGF10 Signaling in Pulmonary Vascular Formation
Fibroblast growth factor 10 (FGF10)is one of the most important genes expressed during lung development. Extensive research has shown that it is expressed in the submesothelial mesenchyme [83][84]. After secretion, FGF10 acts in a paracrine fashion mainly through the receptor FGFR2b located on the lung epithelium [85]. More and more research findings which have mainly used preclinical mouse models for lung injury, have confirmed the role of FGF10 in protecting and regenerating the lung tissue during and after lung injury, e.g., in lung fibrosis [86][87][88]. Our recent data emphasized, that FGF10 indeed plays a crucial role in branching morphogenesis and formation of the pulmonary vasculature during development [89]. As previously mentioned, the temporal–spatial proximity of the lung vasculature and the distal epithelium suggests a close interaction between these two important structures via endothelial–epithelial tissue crosstalk. Thus, it has been shown that mesenchymally secreted FGF10 leads to the upregulation of VEGF in the distal epithelium [90]. Also supporting this hypothesis, we demonstrated, fifteen years ago, that treatment of embryonic lung explants with recombinant vascular endothelial growth factor A (Vegfa) not only upregulates Vegfr2 in the mesenchyme but also induces branching of the epithelium [91]. This finding underscores the notion, that branching morphogenesis and pulmonary vascular formation occur in a highly coordinated fashion. Interestingly, isolated lung epithelium treated in vitrowith VEGF does not respond by branching indicating that the effect of VEGFA on epithelial branching is likely mediated via its action on the mesenchyme. This endothelial–epithelial tissue crosstalk has been extensively investigated by using in vitrostudies co-culturing epithelium with the mesenchyme as well as by in vivolung agenesis model . By inducing the expression of a soluble dominant negative receptor of VEGFR1, it has been shown that Sprouty2 (Spry2), a negative regulator of tyrosine kinase signaling, is upregulated in the epithelium. This suggests inhibition of FGF signaling . Further evidence of the endothelial–epithelial crosstalk has been demonstrated convincingly by using mice with genetic deletion of Pecam1. These mice could neither form endothelial cells nor undergo alveologenesis. Similarly, mice owning a hypomorphic allele for Fgf10display decreased expression of Vegfaand Pecam resulting in simplified lung structure accompanied by an abnormally developed lung vasculature [92].
Our follow-up study on these Fgf10 hypomorphic mice showed that Fgf10 heterozygous newborn mice exposed to hyperoxia (85% hyperoxia, P0–P8) displayed a BPD-like lung phenotype with increased hypoalveolarization accompanied with decreased number of AT2 but an increased number of ATI. Further, transcriptomic analysis of isolated AT2 cells from mutant vs. control lungs revealed downregulated AT2 but upregulated AT1 gene signature in mutant AT2 cells associated with decreased surfactant protein b and c production. AT2 cells could, therefore, be both quantitatively and qualitatively dysfunctional in Fgf10 hypomorphic lungs and it is possible that this could affect the proper formation of the pulmonary vasculature leading to PH in BPD[89].
Intriguingly, investigations on lung tissue of preterm infants that died from severe BPD revealed decreased FGF10 expression [93]. While the direct effect of FGF10 on the lung endothelium still needs to be proven, the response of endothelial cells (ECs) to FGF signaling has been well described in vitro[94][95]. It has been shown by gene expression analysis that FGFR1 and FGFR2 are the major FGF receptors being expressed by ECs while FGFR3seems to play a minor role [96][97].FGFR4 was not found to be expressed in ECs [98]. The functional role of FGF signaling has been demonstrated by using a dominant negative receptor of all FGFRs (FGFR1DN). Murakami et al. showed that FGF signaling was important for homeostasis and vascular integrity [99][100][101]. The same workers showed that FGF signaling acted upstream of VEGF signaling in normal and disease conditions [98][100]. In order to test the cell-specific role of FGFR1/2 in ECs, Oladipupo and colleagues used genetic modified mice to delete FGFR1/2 in Flk1 and Tie2 expressing cells [102]. Both are cell markers for endothelial and hematopoietic cells. They showed that the genetic deletion of Fgfr1/2 in endothelial cells did not lead to disruption of embryonic vascular development. In the adult mutant mice, the vascular integrity was maintained in the homeostatic condition. However, in injury models of eye and skin injury, the neovascularization process was significantly impaired and associated with delayed wound healing. Further evidence for the importance of FGFR1/2 response in ECs upon injury was brought by House and colleagues. Again, using the Tie2-Cre;Fgfr1flox/flox;Fgfr2flox/floxmice, they demonstrated a worsened cardiac function (decreased vessel density, increased endothelial cell apoptosis, and hypokinetic areas) following cardiac ischemia-reperfusion injury [103]. The ligand acting on the endothelial cells via Fgfr1 and Fgfr2 is still unknown. Whether Fgf10 is acting directly on the endothelium or on the VSMCs still needs to be tested.
It has been shown in mice that Fgf10-positive cells were progenitors for VSMCs in vivoduring lung development. The genetically modified mice displaying a knock in of the CreERT2 frame with the first exon of FGF10 crossed with the tdTomato reporter (FGF10iCre/+; tomatoflox/+) were used. These mice allowed us to permanently label Fgf10-positive cells following tamoxifen treatment. Single IP injection of tamoxifen was carried out at E11.5 or E15.5 and embryonic lungs were harvested at E18.5. Two waves of Fgf10 expression during embryonic lung development were observed. In the first wave, the Fgf10-positive cells residing in the submesothelial mesenchyme (and their progeny) contributed to the formation of airway smooth muscle cells (ASMCs), VSMCs, and lipofibroblasts at E18.5. Interestingly, the second wave of Fgf10-positive cells labeled at E15.5 did not give rise to VSMCs [104]. Several studies have suggested that the abnormal accumulation of SMCs in the distal pulmonary arterioles leading to its excessive muscularization was a major contributing factor to the pathology of PH. In a mouse model of hypoxia-induced PH, studies have shown that a significant amount of distal arteriole SMCs were derived from pre-existing smooth muscle cells. It could be possible that these SMCs arising from Fgf10-positive cells during early development, could be one of the key cell types responsible for the increased muscularization of distal arterioles during hypoxia-induced PH. However, this needs further confirmation.
In a previous but nevertheless related study, Mlc1v-nLacZ-24transgenic mice (thereafter called Fgf10LacZ), in which the expression of LacZ was controlled by FGF10 regulatory sequences, were used to monitor the localization of Fgf10 positive cells. By using whole-mount in-situ hybridization and X-gal staining on the E12.5 and E13.5 embryonic lungs, it was found that β-galactosidase-positive cells were present exclusively in the distal mesenchyme, as well as adjacent to the bronchial epithelium. In addition, these β-galactosidase-positive cells around the bronchial epithelium also expressed the smooth muscle cell marker α-SMA suggesting that in the distal mesenchyme, the β-galactosidase-positive cells (Fgf10-positive cells) were progenitors for ASMCs. Reciprocal xenotransplantation on wild type and Fgf10LacZ/+embryos of the distal mesenchyme, as well as live imaging, demonstrated that β-galactosidase-positive cells in the distal mesenchyme were passively relocated around the bronchi [84].
In further studies, it was demonstrated that the Mlc1v-nLacZ-24allele behaved as an hypomorphic Fgf10 allele opening the way to generate allelic series of mice displaying different levels of FGF10 expression. These allelic series were generated by crossing the Fgf10LacZ transgenic line with Fgf10+/– mice on a C57Bl/6 background. Controls (Fgf10+/+) and Fgf10LacZ/– embryos were generated at different developmental stages.
By performing whole-mount immunohistochemistry with α-SMA antibodies in control and Fgf10LacZ/-, theauthorsfound that the reduction in Fgf10 expression led to decreased SMCs around bronchi, as well as a completely disorganized vasculature correlating with decreased VEGF expression.
Besides Fgf10 signaling, other FGFs are also involved in the formation of the pulmonary vasculature. White et al. demonstrated that both fibroblast growth factor 9 (Fgf9) and sonic hedgehog (SHH) signaling to the pulmonary mesenchyme (not the endothelial cells) was necessary and sufficient for the development of distal capillaries. However, Fgf9 could only partially reverse the decreased capillary density found without SHH signaling. SHH could not rescue the vascular phenotype of Fgf9null lungs. These results suggest that Fgf9 and SHH signaling synergistically regulate the growth and pattern of the pulmonary capillary plexus and regulate the temporal and spatial expression of Vegfa .FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network.
4. Conclusions
In conclusion, BPD-PH is affecting a population of preterm infants which should not be neglected, with considerably high burdens for the patients and the health care system. On the basis of data mainly gained from preclinical research in mice, it has become increasingly evident that vascular remodeling (VR) is the pathological basis for pulmonary hypertension in BPD.
However, the origin of the newly formed VSMCs during VR (in both the proximal arteries and the more distal arterioles) and their fate during reverse remodeling is not yet understood. In the future, lineage tracing experiments in the context of experimental BPD in mice (using the Sugen inhibitor 5416 to block VEGF signaling) will be instrumental to characterize the progenitors for these newly formed VSMCs in the disease context. Targeting these progenitor cells could provide an opportunity to treat the disease in human patients.
The BPD mouse model is a well-established and widely accepted model to mimicthe lung phenotype of BPD (alveolar simplification and dysmorphic lung vasculature) but in the context of BPD-PH, it needs to be mentioned that the only use of hyperoxia to induce lung injury is not causing PH in mice. A BPD-PH animal model needs to be established. It has been shown recently, that blockade of FGFR2b ligands activity postnatally in a BPD mouse model lead to decreased blood vessel number and increased muscularization resembling a PH-like phenotype. Unpublished data also suggests that mice with constitutive FGF10/FGFR2b signaling deficiency (Fgf10+/-, Fgfr2b+/-) in room air are more susceptible to develop PH during aging as compared with wildtype mice.
FGF10 and FGFR2b signaling is believed to be essential in lung development. More interestingly, growing evidence coming from experimental research is confirming its preventive and regenerative effect during regeneration in lung diseases, such as BPD. In line with this notion, clinical-grade recombinant FGF10 could have therapeutic potential for the treatment of BPD-PH. However, based on the facts that other FGFs (e.g., FGF1 and FGF2) have been shown to be involved in the remodeling process of bronchial airways in COPD, thus, FGFs are potentially detrimental. Therefore, the therapeutic use of FGF10 needs to be thoroughly validated [105]. In addition, potential adverse side effects need to be evaluated [106]. As a first translational step, the authors suggest the validation of the preventive and therapeutic effect of recombinant FGF10 in a mouse model of BPD-PH. Large animal models would allow the intratracheal or inhalative administration of recombinant FGF10 which would be much more favorable and clinically relevant.
Apart from the abovementioned translational approaches, FGF10 expression (gene and protein levels) needs to be better clinically characterized within the heterogeneous cohort of preterm infants at risk of developing BPD-PH (e.g., gestation age, risk factors for BPD-PH, postnatal ventilation regimes, and genetic predisposition). Thus, future studies including the measurement, for example, using tracheal aspirates or exhaled breath condensate (EBC), of the expression of FGF10 in preterm infants is highly desirable. The establishment of a centralized biobank and databank would promote collection and characterization of biomaterials, provide organized availability and facilitate accessibility for research. In parallel to efforts invested in the clinical setting, more basic research needs to be done in order to gain deeper insight into the cellular and molecular mechanisms of FGF signaling governing vascular remodeling and reverse remodeling processes in BPD-PH.
The combination of both clinical and basic research would allow a better design of therapeutic strategies to prevent and to treat BPD-PH in the future.
References
- Blencowe, H.; Cousens, S.; Oestergaard, M.Z.; Chou, D.; Moller, A.B.; Narwal, R.; Adler, A.; Vera Garcia, C.; Rohde, S.; Say, L.; et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet 2012, 379, 2162–2172.
- Chawanpaiboon, S.; Vogel, J.P.; Moller, A.B.; Lumbiganon, P.; Petzold, M.; Hogan, D.; Landoulsi, S.; Jampathong, N.; Kongwattanakul, K.; Laopaiboon, M.; et al. Global, regional, and national estimates of levels of preterm birth in 2014: A systematic review and modelling analysis. Lancet Glob. Health 2019, 7, e37–e46.
- Stoll, B.J.; Hansen, N.I.; Bell, E.F.; Walsh, M.C.; Carlo, W.A.; Shankaran, S.; Laptook, A.R.; Sanchez, P.J.; Van Meurs, K.P.; Wyckoff, M.; et al. Trends in Care Practices, Morbidity, and Mortality of Extremely Preterm Neonates, 1993–2012. JAMA 2015, 314, 1039–1051.
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980.
- Popova, A.P.; Bentley, J.K.; Cui, T.X.; Richardson, M.N.; Linn, M.J.; Lei, J.; Chen, Q.; Goldsmith, A.M.; Pryhuber, G.S.; Hershenson, M.B. Reduced platelet-derived growth factor receptor expression is a primary feature of human bronchopulmonary dysplasia. Am. J. Physiol. Lung C 2014, 307, L231–L239.
- Speer, C.P. Inflammation and bronchopulmonary dysplasia: A continuing story. Semin. Fetal Neonat. Med. 2006, 11, 354–362.
- Al-Ghanem, G.; Shah, P.; Thomas, S.; Banfield, L.; El Helou, S.; Fusch, C.; Mukerji, A. Bronchopulmonary dysplasia and pulmonary hypertension: A meta-analysis. J. Perinatol. 2017, 37, 414–419.
- Arjaans, S.; Zwart, E.A.H.; Ploegstra, M.J.; Bos, A.F.; Kooi, E.M.W.; Hillege, H.L.; Berger, R.M.F. Identification of gaps in the current knowledge on pulmonary hypertension in extremely preterm infants: A systematic review and meta-analysis. Paediatr. Perinat. Epidemiol. 2018, 32, 258–267.
- Sheikh, A.Q.; Lighthouse, J.K.; Greif, D.M. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 2014, 6, 809–817.
- Shimoda, L.A.; Laurie, S.S. Vascular remodeling in pulmonary hypertension. J. Mol. Med. Berl. 2013, 91, 297–309.
- Tan, W.; Madhavan, K.; Hunter, K.S.; Park, D.; Stenmark, K.R. Vascular stiffening in pulmonary hypertension: Cause or consequence? (2013 Grover Conference series). Pulm. Circ. 2014, 4, 560–580.
- Frank, D.B.; Crystal, M.A.; Morales, D.L.; Gerald, K.; Hanna, B.D.; Mallory, G.B., Jr.; Rossano, J.W. Trends in pediatric pulmonary hypertension-related hospitalizations in the United States from 2000–2009. Pulm. Circ. 2015, 5, 339–348.
- Haworth, S.G.; Hislop, A.A. Treatment and survival in children with pulmonary arterial hypertension: The UK Pulmonary Hypertension Service for Children 2001–2006. Heart 2009, 95, 312–317.
- Abman, S.H.; Hansmann, G.; Archer, S.L.; Ivy, D.D.; Adatia, I.; Chung, W.K.; Hanna, B.D.; Rosenzweig, E.B.; Raj, J.U.; Cornfield, D.; et al. Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation 2015, 132, 2037–2099.
- Krishnan, U.; Feinstein, J.A.; Adatia, I.; Austin, E.D.; Mullen, M.P.; Hopper, R.K.; Hanna, B.; Romer, L.; Keller, R.L.; Fineman, J.; et al. Evaluation and Management of Pulmonary Hypertension in Children with Bronchopulmonary Dysplasia. J. Pediatr. 2017, 188, 24–34.
- Hansmann, G.; Apitz, C.; Abdul-Khaliq, H.; Alastalo, T.P.; Beerbaum, P.; Bonnet, D.; Dubowy, K.O.; Gorenflo, M.; Hager, A.; Hilgendorff, A.; et al. Executive summary. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102 (Suppl. S2), ii86–ii100.
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41, doi:10.1016/j.jacc.2013.10.029.
- Ezekian, J.E.; Hill, K.D. Management of Pulmonary Arterial Hypertension in the Pediatric Patient. Curr. Cardiol. Rep. 2019, 21, 162, doi:10.1007/s11886-019-1229-2.
- Thompson, A.A.R.; Lawrie, A. Targeting Vascular Remodeling to Treat Pulmonary Arterial Hypertension. Trends Mol. Med. 2017, 23, 31–45, doi:10.1016/j.molmed.2016.11.005.
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M.F. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 2019, 53, doi:10.1183/13993003.01916-2018.
- Mammoto, A.; Mammoto, T. Vascular Niche in Lung Alveolar Development, Homeostasis, and Regeneration. Front. Bioeng. Biotechnol. 2019, 7, 318, doi:10.3389/fbioe.2019.00318.
- Chao, C.M.; El Agha, E.; Tiozzo, C.; Minoo, P.; Bellusci, S. A breath of fresh air on the mesenchyme: Impact of impaired mesenchymal development on the pathogenesis of bronchopulmonary dysplasia. Front. Med. Lausanne 2015, 2, 27, doi:10.3389/fmed.2015.00027.
- Gebb, S.A.; Shannon, J.M. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev. Dyn. 2000, 217, 159–169, doi:10.1002/(SICI)1097-0177(200002)217:2<159::AID-DVDY3>3.0.CO;2-9.
- Schittny, J.C. Development of the lung. Cell Tissue Res. 2017, 367, 427–444, doi:10.1007/s00441-016-2545-0.
- Stenmark, K.R.; Abman, S.H. Lung vascular development: Implications for the pathogenesis of bronchopulmonary dysplasia. Annu. Rev. Physiol. 2005, 67, 623–661, doi:10.1146/annurev.physiol.67.040403.102229.
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front. Cardiovasc. Med. 2018, 5, 154, doi:10.3389/fcvm.2018.00154.
- Schittny, J.C.; Mund, S.I.; Stampanoni, M. Evidence and structural mechanism for late lung alveolarization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L246–L254, doi:10.1152/ajplung.00296.2007.
- Greenberg, J.M.; Thompson, F.Y.; Brooks, S.K.; Shannon, J.M.; McCormick-Shannon, K.; Cameron, J.E.; Mallory, B.P.; Akeson, A.L. Mesenchymal expression of vascular endothelial growth factors D and A defines vascular patterning in developing lung. Dev. Dyn. 2002, 224, 144–153, doi:10.1002/dvdy.10095.
- White, A.C.; Lavine, K.J.; Ornitz, D.M. FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network. Development 2007, 134, 3743–3752, doi:10.1242/dev.004879.
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66, doi:10.1038/376062a0.
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70, doi:10.1038/376066a0.
- Yamamoto, H.; Yun, E.J.; Gerber, H.P.; Ferrara, N.; Whitsett, J.A.; Vu, T.H. Epithelial-Vascular cross talk mediated by VEGF-A and HGF signaling directs primary septae formation during distal lung morphogenesis. Dev. Biol. 2007, 308, 44–53, doi:10.1016/j.ydbio.2007.04.042.
- Zhao, L.; Wang, K.; Ferrara, N.; Vu, T.H. Vascular endothelial growth factor co-ordinates proper development of lung epithelium and vasculature. Mech. Dev. 2005, 122, 877–886, doi:10.1016/j.mod.2005.04.001.
- Yang, J.; Hernandez, B.J.; Martinez Alanis, D.; Narvaez del Pilar, O.; Vila-Ellis, L.; Akiyama, H.; Evans, S.E.; Ostrin, E.J.; Chen, J. The development and plasticity of alveolar type 1 cells. Development 2016, 143, 54–65, doi:10.1242/dev.130005.
- Greif, D.M.; Kumar, M.; Lighthouse, J.K.; Hum, J.; An, A.; Ding, L.; Red-Horse, K.; Espinoza, F.H.; Olson, L.; Offermanns, S.; et al. Radial construction of an arterial wall. Dev. Cell. 2012, 23, 482–493, doi:10.1016/j.devcel.2012.07.009.
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464, doi:10.1215/S1152851705000232.
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell. 2011, 21, 193–215, doi:10.1016/j.devcel.2011.07.001.
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313, doi:10.1016/j.stem.2008.07.003.
- Paiva, A.E.; Lousado, L.; Guerra, D.A.P.; Azevedo, P.O.; Sena, I.F.G.; Andreotti, J.P.; Santos, G.S.P.; Goncalves, R.; Mintz, A.; Birbrair, A. Pericytes in the Premetastatic Niche. Cancer Res. 2018, 78, 2779–2786, doi:10.1158/0008-5472.CAN-17-3883.
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66, doi:10.1016/j.stem.2014.11.004.
- Eilken, H.M.; Dieguez-Hurtado, R.; Schmidt, I.; Nakayama, M.; Jeong, H.W.; Arf, H.; Adams, S.; Ferrara, N.; Adams, R.H. Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat. Commun. 2017, 8, 1574, doi:10.1038/s41467-017-01738-3.
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055.
- Jarad, M.; Kuczynski, E.A.; Morrison, J.; Viloria-Petit, A.M.; Coomber, B.L. Release of endothelial cell associated VEGFR2 during TGF-beta modulated angiogenesis in vitro. BMC Cell Biol. 2017, 18, 10, doi:10.1186/s12860-017-0127-y.
- Wakui, S.; Yokoo, K.; Muto, T.; Suzuki, Y.; Takahashi, H.; Furusato, M.; Hano, H.; Endou, H.; Kanai, Y. Localization of Ang-1, -2, Tie-2, and VEGF expression at endothelial-pericyte interdigitation in rat angiogenesis. Lab. Invest. 2006, 86, 1172–1184, doi:10.1038/labinvest.3700476.
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/Pericyte interactions. Circ. Res. 2005, 97, 512–523, doi:10.1161/01.RES.0000182903.16652.d7.
- Kato, K.; Dieguez-Hurtado, R.; Park, D.Y.; Hong, S.P.; Kato-Azuma, S.; Adams, S.; Stehling, M.; Trappmann, B.; Wrana, J.L.; Koh, G.Y.; et al. Pulmonary pericytes regulate lung morphogenesis. Nat. Commun. 2018, 9, 2448, doi:10.1038/s41467-018-04913-2.
- Yuan, K.; Shamskhou, E.A.; Orcholski, M.E.; Nathan, A.; Reddy, S.; Honda, H.; Mani, V.; Zeng, Y.; Ozen, M.O.; Wang, L.; et al. Loss of Endothelium-Derived Wnt5a Is Associated With Reduced Pericyte Recruitment and Small Vessel Loss in Pulmonary Arterial Hypertension. Circulation 2019, 139, 1710–1724, doi:10.1161/CIRCULATIONAHA.118.037642.
- Shammout, B.; Johnson, J.R. Pericytes in Chronic Lung Disease. Adv. Exp. Med. Biol. 2019, 1147, 299–317, doi:10.1007/978-3-030-16908-4_14.
- Gao, Y.; Chen, T.; Raj, J.U. Endothelial and Smooth Muscle Cell Interactions in the Pathobiology of Pulmonary Hypertension. Am. J. Respir. Cell. Mol. Biol. 2016, 54, 451–460, doi:10.1165/rcmb.2015-0323TR.
- Hadchouel, A.; Durrmeyer, X.; Bouzigon, E.; Incitti, R.; Huusko, J.; Jarreau, P.H.; Lenclen, R.; Demenais, F.; Franco-Montoya, M.L.; Layouni, I.; et al. Identification of SPOCK2 as a susceptibility gene for bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2011, 184, 1164–1170, doi:10.1164/rccm.201103-0548OC.
- Chao, C.M.; Moiseenko, A.; Kosanovic, D.; Rivetti, S.; El Agha, E.; Wilhelm, J.; Kampschulte, M.; Yahya, F.; Ehrhardt, H.; Zimmer, K.P.; et al. Impact of Fgf10 deficiency on pulmonary vasculature formation in a mouse model of bronchopulmonary dysplasia. Hum. Mol. Genet. 2019, 28, 1429–1444, doi:10.1093/hmg/ddy439.
- Yee, M.; White, R.J.; Awad, H.A.; Bates, W.A.; McGrath-Morrow, S.A.; O’Reilly, M.A. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am. J. Pathol. 2011, 178, 2601–2610, doi:10.1016/j.ajpath.2011.02.010.
- Tiozzo, C.; Carraro, G.; Al Alam, D.; Baptista, S.; Danopoulos, S.; Li, A.; Lavarreda-Pearce, M.; Li, C.; De Langhe, S.; Chan, B.; et al. Mesodermal Pten inactivation leads to alveolar capillary dysplasia- like phenotype. J. Clin. Invest. 2012, 122, 3862–3872, doi:10.1172/JCI61334.
- Abman, S.H. Bronchopulmonary dysplasia: A vascular hypothesis. Am. J. Respir. Crit. Care Med. 2001, 164, 1755–1756, doi:10.1164/ajrccm.164.10.2109111c.
- Baker, C.D.; Abman, S.H. Impaired pulmonary vascular development in bronchopulmonary dysplasia. Neonatology 2015, 107, 344–351, doi:10.1159/000381129.
- Greco, F.; Wiegert, S.; Baumann, P.; Wellmann, S.; Pellegrini, G.; Cannizzaro, V. Hyperoxia-Induced lung structure-function relation, vessel rarefaction, and cardiac hypertrophy in an infant rat model. J. Transl. Med. 2019, 17, 91, doi:10.1186/s12967-019-1843-1.
- Crnkovic, S.; Marsh, L.M.; El Agha, E.; Voswinckel, R.; Ghanim, B.; Klepetko, W.; Stacher-Priehse, E.; Olschewski, H.; Bloch, W.; Bellusci, S.; et al. Resident cell lineages are preserved in pulmonary vascular remodeling. J. Pathol. 2018, 244, 485–498, doi:10.1002/path.5044.
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272, doi:10.1164/rccm.201201-0164OC.
- Stenmark, K.R.; Frid, M.; Perros, F. Endothelial-To-Mesenchymal Transition: An Evolving Paradigm and a Promising Therapeutic Target in PAH. Circulation 2016, 133, 1734–1737, doi:10.1161/CIRCULATIONAHA.116.022479.
- Assaad, A.M.; Kawut, S.M.; Arcasoy, S.M.; Rosenzweig, E.B.; Wilt, J.S.; Sonett, J.R.; Borczuk, A.C. Platelet-Derived growth factor is increased in pulmonary capillary hemangiomatosis. Chest 2007, 131, 850–855, doi:10.1378/chest.06-1680.
- Ricard, N.; Tu, L.; Le Hiress, M.; Huertas, A.; Phan, C.; Thuillet, R.; Sattler, C.; Fadel, E.; Seferian, A.; Montani, D.; et al. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation 2014, 129, 1586–1597, doi:10.1161/CIRCULATIONAHA.113.007469.
- Lin, S.L.; Chang, F.C.; Schrimpf, C.; Chen, Y.T.; Wu, C.F.; Wu, V.C.; Chiang, W.C.; Kuhnert, F.; Kuo, C.J.; Chen, Y.M.; et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am. J. Pathol. 2011, 178, 911–923, doi:10.1016/j.ajpath.2010.10.012.
- Wang, Y.C.; Chen, Q.; Luo, J.M.; Nie, J.; Meng, Q.H.; Shuai, W.; Xie, H.; Xia, J.M.; Wang, H. Notch1 promotes the pericyte-myofibroblast transition in idiopathic pulmonary fibrosis through the PDGFR/ROCK1 signal pathway. Exp. Mol. Med. 2019, 51, 1–11, doi:10.1038/s12276-019-0228-0.
- Li, C.; Lee, M.K.; Gao, F.; Webster, S.; Di, H.; Duan, J.; Yang, C.Y.; Bhopal, N.; Peinado, N.; Pryhuber, G.; et al. Secondary crest myofibroblast PDGFRalpha controls the elastogenesis pathway via a secondary tier of signaling networks during alveologenesis. Development 2019, 146, doi:10.1242/dev.176354.
- Oak, P.; Pritzke, T.; Thiel, I.; Koschlig, M.; Mous, D.S.; Windhorst, A.; Jain, N.; Eickelberg, O.; Foerster, K.; Schulze, A.; et al. Attenuated PDGF signaling drives alveolar and microvascular defects in neonatal chronic lung disease. EMBO Mol. Med. 2017, 9, 1504–1520, doi:10.15252/emmm.201607308.
- Xing, Y.; Fu, J.; Yang, H.; Yao, L.; Qiao, L.; Du, Y.; Xue, X. MicroRNA expression profiles and target prediction in neonatal Wistar rat lungs during the development of bronchopulmonary dysplasia. Int. J. Mol. Med. 2015, 36, 1253–1263, doi:10.3892/ijmm.2015.2347.
- Yang, Y.; Qiu, J.; Kan, Q.; Zhou, X.G.; Zhou, X.Y. MicroRNA expression profiling studies on bronchopulmonary dysplasia: A systematic review and meta-analysis. Genet. Mol. Res. 2013, 12, 5195–5206, doi:10.4238/2013.October.30.4.
- Yang, S.; Banerjee, S.; Freitas, A.; Cui, H.; Xie, N.; Abraham, E.; Liu, G. miR-21 regulates chronic hypoxia-induced pulmonary vascular remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L521–L529, doi:10.1152/ajplung.00316.2011.
- Bhaskaran, M.; Xi, D.; Wang, Y.; Huang, C.; Narasaraju, T.; Shu, W.; Zhao, C.; Xiao, X.; More, S.; Breshears, M.; et al. Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiol. Genomics 2012, 44, 970–980, doi:10.1152/physiolgenomics.00145.2011.
- Chen, X.; Talati, M.; Fessel, J.P.; Hemnes, A.R.; Gladson, S.; French, J.; Shay, S.; Trammell, A.; Phillips, J.A.; Hamid, R.; et al. Estrogen Metabolite 16alpha-Hydroxyestrone Exacerbates Bone Morphogenetic Protein Receptor Type II-Associated Pulmonary Arterial Hypertension Through MicroRNA-29-Mediated Modulation of Cellular Metabolism. Circulation 2016, 133, 82–97, doi:10.1161/CIRCULATIONAHA.115.016133.
- Hu, Y.; Xie, L.; Yu, J.; Fu, H.; Zhou, D.; Liu, H. Inhibition of microRNA-29a alleviates hyperoxia-induced bronchopulmonary dysplasia in neonatal mice via upregulation of GAB1. Mol. Med. 2019, 26, 3, doi:10.1186/s10020-019-0127-9.
- Dong, J.; Carey, W.A.; Abel, S.; Collura, C.; Jiang, G.; Tomaszek, S.; Sutor, S.; Roden, A.C.; Asmann, Y.W.; Prakash, Y.S.; et al. MicroRNA-mRNA interactions in a murine model of hyperoxia-induced bronchopulmonary dysplasia. BMC Genomics 2012, 13, 204, doi:10.1186/1471-2164-13-204.
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173, doi:10.1038/s41467-017-01349-y.
- Ruiz-Camp, J.; Quantius, J.; Lignelli, E.; Arndt, P.F.; Palumbo, F.; Nardiello, C.; Surate Solaligue, D.E.; Sakkas, E.; Mizikova, I.; Rodriguez-Castillo, J.A.; et al. Targeting miR-34a/Pdgfra interactions partially corrects alveologenesis in experimental bronchopulmonary dysplasia. EMBO Mol. Med. 2019, 11, doi:10.15252/emmm.201809448.
- Potus, F.; Ruffenach, G.; Dahou, A.; Thebault, C.; Breuils-Bonnet, S.; Tremblay, E.; Nadeau, V.; Paradis, R.; Graydon, C.; Wong, R.; et al. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation 2015, 132, 932–943, doi:10.1161/CIRCULATIONAHA.115.016382.
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032, doi:10.1016/j.celrep.2015.09.049.
- Chao, C.M.; Carraro, G.; Rako, Z.A.; Kolck, J.; Sedighi, J.; Zimmermann, V.; Moiseenko, A.; Wilhelm, J.; Young, B.M.; Chong, L.; et al. Failure to Down-Regulate miR-154 Expression in Early Postnatal Mouse Lung Epithelium Suppresses Alveologenesis, with Changes in Tgf-beta Signaling Similar to those Induced by Exposure to Hyperoxia. Cells 2020, 9, doi:10.3390/cells9040859.
- Bernardo, B.C.; Nguyen, S.S.; Gao, X.M.; Tham, Y.K.; Ooi, J.Y.; Patterson, N.L.; Kiriazis, H.; Su, Y.; Thomas, C.J.; Lin, R.C.; et al. Inhibition of miR-154 Protects Against Cardiac Dysfunction and Fibrosis in a Mouse Model of Pressure Overload. Sci. Rep. 2016, 6, 22442, doi:10.1038/srep22442.
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887, doi:10.1165/rcmb.2011-0377OC.
- Yuan, H.S.; Xiong, D.Q.; Huang, F.; Cui, J.; Luo, H. MicroRNA-421 inhibition alleviates bronchopulmonary dysplasia in a mouse model via targeting Fgf10. J. Cell. Biochem. 2019, 120, 16876–16887, doi:10.1002/jcb.28945.
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82, doi:10.1038/nm.3040.
- Chen, J.; Cui, X.; Li, L.; Qu, J.; Raj, J.U.; Gou, D. MiR-339 inhibits proliferation of pulmonary artery smooth muscle cell by targeting FGF signaling. Physiol. Rep. 2017, 5, doi:10.14814/phy2.13441.
- Jones, M.R.; Lingampally, A.; Dilai, S.; Shrestha, A.; Stripp, B.; Helmbacher, F.; Chen, C.; Chao, C.M.; Bellusci, S. Characterization of Tg(Etv4-GFP) and Etv5 (RFP) Reporter Lines in the Context of Fibroblast Growth Factor 10 Signaling During Mouse Embryonic Lung Development. Front. Genet. 2019, 10, 178, doi:10.3389/fgene.2019.00178.
- Mailleux, A.A.; Kelly, R.; Veltmaat, J.M.; De Langhe, S.P.; Zaffran, S.; Thiery, J.P.; Bellusci, S. Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development 2005, 132, 2157–2166, doi:10.1242/dev.01795.
- Ohuchi, H.; Hori, Y.; Yamasaki, M.; Harada, H.; Sekine, K.; Kato, S.; Itoh, N. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem. Biophys. Res. Commun. 2000, 277, 643–649, doi:10.1006/bbrc.2000.3721.
- Gupte, V.V.; Ramasamy, S.K.; Reddy, R.; Lee, J.; Weinreb, P.H.; Violette, S.M.; Guenther, A.; Warburton, D.; Driscoll, B.; Minoo, P.; et al. Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care. Med. 2009, 180, 424–436, doi:10.1164/rccm.200811-1794OC.
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S.P. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Invest. 2011, 121, 4409–4419, doi:10.1172/JCI58097.
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273, doi:10.1016/j.stem.2016.10.004.
- Chao, C.M.; Yahya, F.; Moiseenko, A.; Tiozzo, C.; Shrestha, A.; Ahmadvand, N.; El Agha, E.; Quantius, J.; Dilai, S.; Kheirollahi, V.; et al. Fgf10 deficiency is causative for lethality in a mouse model of bronchopulmonary dysplasia. J. Pathol. 2017, 241, 91–103, doi:10.1002/path.4834.
- Scott, C.L.; Walker, D.J.; Cwiklinski, E.; Tait, C.; Tee, A.R.; Land, S.C. Control of HIF-1{alpha} and vascular signaling in fetal lung involves cross talk between mTORC1 and the FGF-10/FGFR2b/Spry2 airway branching periodicity clock. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L455–L471, doi:10.1152/ajplung.00348.2009.
- Del Moral, P.M.; Sala, F.G.; Tefft, D.; Shi, W.; Keshet, E.; Bellusci, S.; Warburton, D. VEGF-A signaling through Flk-1 is a critical facilitator of early embryonic lung epithelial to endothelial crosstalk and branching morphogenesis. Dev. Biol. 2006, 290, 177–188, doi:10.1016/j.ydbio.2005.11.022.
- Ramasamy, S.K.; Mailleux, A.A.; Gupte, V.V.; Mata, F.; Sala, F.G.; Veltmaat, J.M.; Del Moral, P.M.; De Langhe, S.; Parsa, S.; Kelly, L.K.; et al. Fgf10 dosage is critical for the amplification of epithelial cell progenitors and for the formation of multiple mesenchymal lineages during lung development. Dev. Biol. 2007, 307, 237–247, doi:10.1016/j.ydbio.2007.04.033.
- Benjamin, J.T.; Smith, R.J.; Halloran, B.A.; Day, T.J.; Kelly, D.R.; Prince, L.S. FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by Toll-like receptor activation. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2007, 292, L550–L558, doi:10.1152/ajplung.00329.2006.
- Seghezzi, G.; Patel, S.; Ren, C.J.; Gualandris, A.; Pintucci, G.; Robbins, E.S.; Shapiro, R.L.; Galloway, A.C.; Rifkin, D.B.; Mignatti, P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An autocrine mechanism contributing to angiogenesis. J. Cell Biol. 1998, 141, 1659–1673, doi:10.1083/jcb.141.7.1659.
- Gospodarowicz, D.; Brown, K.D.; Birdwell, C.R.; Zetter, B.R. Control of proliferation of human vascular endothelial cells. Characterization of the response of human umbilical vein endothelial cells to fibroblast growth factor, epidermal growth factor, and thrombin. J. Cell Biol. 1978, 77, 774–788, doi:10.1083/jcb.77.3.774.
- Claus, P.; Grothe, C. Molecular cloning and developmental expression of rat fibroblast growth factor receptor 3. Histochem. Cell Biol. 2001, 115, 147–155, doi:10.1007/s004180000215.
- Shin, J.W.; Min, M.; Larrieu-Lahargue, F.; Canron, X.; Kunstfeld, R.; Nguyen, L.; Henderson, J.E.; Bikfalvi, A.; Detmar, M.; Hong, Y.K. Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: A role for FGF signaling in lymphangiogenesis. Mol. Biol. Cell 2006, 17, 576–584, doi:10.1091/mbc.e05-04-0368.
- Lieu, C.; Heymach, J.; Overman, M.; Tran, H.; Kopetz, S. Beyond VEGF: Inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin. Cancer Res. 2011, 17, 6130–6139, doi:10.1158/1078-0432.CCR-11-0659.
- Murakami, M.; Elfenbein, A.; Simons, M. Non-Canonical fibroblast growth factor signalling in angiogenesis. Cardiovasc. Res. 2008, 78, 223–231, doi:10.1093/cvr/cvm086.
- Murakami, M.; Nguyen, L.T.; Hatanaka, K.; Schachterle, W.; Chen, P.Y.; Zhuang, Z.W.; Black, B.L.; Simons, M. FGF-Dependent regulation of VEGF receptor 2 expression in mice. J. Clin. Invest. 2011, 121, 2668–2678, doi:10.1172/JCI44762.
- Hatanaka, K.; Lanahan, A.A.; Murakami, M.; Simons, M. Fibroblast growth factor signaling potentiates VE-cadherin stability at adherens junctions by regulating SHP2. PLoS ONE 2012, 7, e37600, doi:10.1371/journal.pone.0037600.
- Oladipupo, S.S.; Smith, C.; Santeford, A.; Park, C.; Sene, A.; Wiley, L.A.; Osei-Owusu, P.; Hsu, J.; Zapata, N.; Liu, F.; et al. Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 13379–13384, doi:10.1073/pnas.1324235111.
- House, S.L.; Castro, A.M.; Lupu, T.S.; Weinheimer, C.; Smith, C.; Kovacs, A.; Ornitz, D.M. Endothelial fibroblast growth factor receptor signaling is required for vascular remodeling following cardiac ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H559–H571, doi:10.1152/ajpheart.00758.2015.
- El Agha, E.; Herold, S.; Al Alam, D.; Quantius, J.; MacKenzie, B.; Carraro, G.; Moiseenko, A.; Chao, C.M.; Minoo, P.; Seeger, W.; et al. Fgf10-Positive cells represent a progenitor cell population during lung development and postnatally. Development 2014, 141, 296–306.
- Kranenburg, A.R.; Willems-Widyastuti, A.; Mooi, W.J.; Saxena, P.R.; Sterk, P.J.; de Boer, W.I.; Sharma, H.S. Chronic obstructive pulmonary disease is associated with enhanced bronchial expression of FGF-1, FGF-2, and FGFR-1. J. Pathol. 2005, 206, 28–38.
- McAuley, D.F.; Cross, L.M.; Hamid, U.; Gardner, E.; Elborn, J.S.; Cullen, K.M.; Dushianthan, A.; Grocott, M.P.; Matthay, M.A.; O’Kane, C.M. Keratinocyte growth factor for the treatment of the acute respiratory distress syndrome (KARE): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Respir. Med. 2017, 5, 484–491

