Bronchopulmonary dysplasia (BPD) is a chronic lung disease affecting preterm infants. More than 50 years after the first description of BPD by Northway, this chronic lung disease affecting many preterm infants is still less understood. Additonally, approximately 40% of preterm infants suffering from severe BPD also suffer from Bronchopulmonary dysplasia-associated pulmonary hypertension (BPD-PH), leading to a significant increase in total morbidity and mortality. Until today, there is no curative therapy for both BPD and BPD-PH available. It has become increasingly evident that growth factors are playing a central role in normal and pathologic development of the pulmonary vasculature. Thus, this review aims to summarize the recent evidence in our understanding of BPD-PH from a basic scientific point of view, focusing on the potential role of Fibroblast Growth Factor (FGF)/FGF10 signaling pathway contributing to disease development, progression and resolution.
- Bronchopulmonary dysplasia
- Pulmonary hypertension
- Fgf signaling
Dear author, the following contents are excerpts from your papers. They are editable.
(Due to the lack of relevant professional knowledge, our editors cannot complete a complete entry by summarizing your paper, so if you are interested in this work. you may need to write some contents by yourself. A good entry will better present your ideas, research and results to other scholars. Readers will also be able to access your paper directly through entries.)
1. Introduction
2. Development of Normal Pulmonary Vasculature and BPD-PH from the Basic Scientific Point-of-View
2.1. Normal Development of Pulmonary Vasculature
2.1.1. Lung Vasculature Formation and Maturation
2.1.2. Pulmonary Vascular Development Needs Multicellular Crosstalks
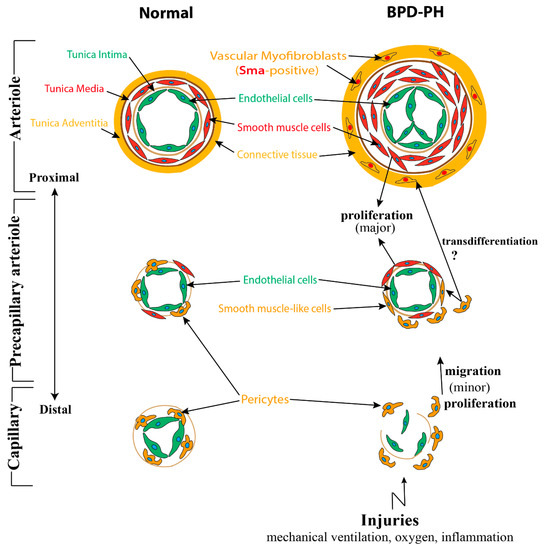
2.2. Pathologic Development of Pulmonary Vasculature Leading to PH in BPD
2.2.1. Abnormalities of Pulmonary Vasculature Observed in BPD-PH
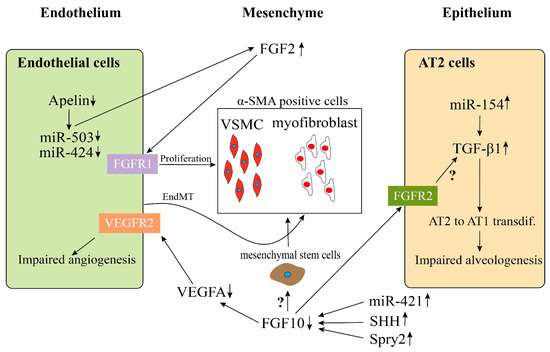
2.2.2. Impaired Multicellular Interactions Disrupt Pulmonary Vascular Development in BPD-PH
2.3. MicroRNAs (miRs) May Be Associated with the Pathologic Vascular Development of BPD-PH
| miRs Shown to be Involved | Changes in PH and BPD | Potential Roles in BPD-PH | References |
|---|---|---|---|
| miR-21 | increased | Increases PASMCs proliferation through inhibiting Bmpr2 activation | [100,101] |
| miR-29 | increased | Impairs PASMCs and ECs function through down-regulating PPARγ | [102,103,104] |
| miR-34a | increased | increases lung epithelial cell apoptosis through down-regulating ANG1-TIE2 signaling; impairs alveologenesis through increasing PDGFR alpha-expressing myofibroblasts | [105,106] |
| miR-126 | decreased | Decreases microvessel density through up-regulating SPRED-1 | [101,107] |
| miR-130 | increased | Promotes ECM remodeling, increases PASMCs proliferation and crosstalk through down-regulating PPARγ-APOE-LRP8 axis | [96,108] |
| miR-154 | increased | Impairs alveologenesis through increasing TGFβ. | [109,110,111] |
| miR-421 | increased | Impairs alveologenesis through down-regulation of FGF10 signaling | [99] |
| miR-503 | decreased | Impairs ECs function and induces PASMCs proliferation through upregulating FGF2 and FGFR1 | [96,98] |
3. Role of FGF/FGF10 Signaling in Pulmonary Vascular Formation
Fibroblast growth factor 10 (FGF10)is one of the most important genes expressed during lung development. Extensive research has shown that it is expressed in the submesothelial mesenchyme [112,113]. After secretion, FGF10 acts in a paracrine fashion mainly through the receptor FGFR2b located on the lung epithelium [114]. More and more research findings which have mainly used preclinical mouse models for lung injury, have confirmed the role of FGF10 in protecting and regenerating the lung tissue during and after lung injury, e.g., in lung fibrosis [115–117]. Our recent data emphasized, that FGF10 indeed plays a crucial role in branching morphogenesis and formation of the pulmonary vasculature during development [79,118]. As previously mentioned, the temporal–spatial proximity of the lung vasculature and the distal epithelium suggests a close interaction between these two important structures via endothelial–epithelial tissue crosstalk. Thus, it has been shown that mesenchymally secreted FGF10 leads to the upregulation of VEGF in the distal epithelium [119]. Also supporting this hypothesis, we demonstrated, fifteen years ago, that treatment of embryonic lung explants with recombinant vascular endothelial growth factor A (Vegfa) not only upregulates Vegfr2 in the mesenchyme but also induces branching of the epithelium [120]. This finding underscores the notion, that branching morphogenesis and pulmonary vascular formation occur in a highly coordinated fashion. Interestingly, isolated lung epithelium treated in vitrowith VEGF does not respond by branching indicating that the effect of VEGFA on epithelial branching is likely mediated via its action on the mesenchyme. This endothelial–epithelial tissue crosstalk has been extensively investigated by using in vitrostudies co-culturing epithelium with the mesenchyme as well as by in vivolung agenesis model [48]. By inducing the expression of a soluble dominant negative receptor of VEGFR1, it has been shown that Sprouty2 (Spry2), a negative regulator of tyrosine kinase signaling, is upregulated in the epithelium. This suggests inhibition of FGF signaling [22]. Further evidence of the endothelial–epithelial crosstalk has been demonstrated convincingly by using mice with genetic deletion of Pecam1. These mice could neither form endothelial cells nor undergo alveologenesis [89]. Similarly, mice owning a hypomorphic allele for Fgf10display decreased expression of Vegfaand Pecam resulting in simplified lung structure accompanied by an abnormally developed lung vasculature [121].
Our follow-up study on these Fgf10 hypomorphic mice showed that Fgf10 heterozygous newborn mice exposed to hyperoxia (85% hyperoxia, P0–P8) displayed a BPD-like lung phenotype with increased hypoalveolarization accompanied with decreased number of AT2 but an increased number of ATI. Further, transcriptomic analysis of isolated AT2 cells from mutant vs. control lungs revealed downregulated AT2 but upregulated AT1 gene signature in mutant AT2 cells associated with decreased surfactant protein b and c production. AT2 cells could, therefore, be both quantitatively and qualitatively dysfunctional in Fgf10 hypomorphic lungs and it is possible that this could affect the proper formation of the pulmonary vasculature leading to PH in BPD [118].
Intriguingly, investigations on lung tissue of preterm infants that died from severe BPD revealed decreased FGF10 expression [122]. While the direct effect of FGF10 on the lung endothelium still needs to be proven, the response of endothelial cells (ECs) to FGF signaling has been well described in vitro[123,124]. It has been shown by gene expression analysis that FGFR1 and FGFR2 are the major FGF receptors being expressed by ECs while FGFR3seems to play a minor role [125,126].FGFR4 was not found to be expressed in ECs [127]. The functional role of FGF signaling has been demonstrated by using a dominant negative receptor of all FGFRs (FGFR1DN). Murakami et al. showed that FGF signaling was important for homeostasis and vascular integrity [128–130]. The same workers showed that FGF signaling acted upstream of VEGF signaling in normal and disease conditions [127,129]. In order to test the cell-specific role of FGFR1/2 in ECs, Oladipupo and colleagues used genetic modified mice to delete FGFR1/2 in Flk1 and Tie2 expressing cells [131]. Both are cell markers for endothelial and hematopoietic cells. They showed that the genetic deletion of Fgfr1/2 in endothelial cells did not lead to disruption of embryonic vascular development. In the adult mutant mice, the vascular integrity was maintained in the homeostatic condition. However, in injury models of eye and skin injury, the neovascularization process was significantly impaired and associated with delayed wound healing. Further evidence for the importance of FGFR1/2 response in ECs upon injury was brought by House and colleagues. Again, using the Tie2-Cre;Fgfr1flox/flox;Fgfr2flox/floxmice, they demonstrated a worsened cardiac function (decreased vessel density, increased endothelial cell apoptosis, and hypokinetic areas) following cardiac ischemia-reperfusion injury [132]. The ligand acting on the endothelial cells via Fgfr1 and Fgfr2 is still unknown. Whether Fgf10 is acting directly on the endothelium or on the VSMCs still needs to be tested.
It has been shown in mice that Fgf10-positive cells were progenitors for VSMCs in vivoduring lung development. The genetically modified mice displaying a knock in of the CreERT2 frame with the first exon of FGF10 crossed with the tdTomato reporter mice(FGF10iCre/+; tomatoflox/+) were used. These mice allowed us to permanently label Fgf10-positive cells following tamoxifen treatment. Single IP injection of tamoxifen was carried out at E11.5 or E15.5 and embryonic lungs were harvested at E18.5. Two waves of Fgf10 expression during embryonic lung development were observed. In the first wave, the Fgf10-positive cells residing in the submesothelial mesenchyme (and their progeny) contributed to the formation of airway smooth muscle cells (ASMCs), VSMCs, and lipofibroblasts at E18.5. Interestingly, the second wave of Fgf10-positive cells labeled at E15.5 did not give rise to VSMCs [133]. Several studies have suggested that the abnormal accumulation of SMCs in the distal pulmonary arterioles leading to its excessive muscularization was a major contributing factor to the pathology of PH. In a mouse model of hypoxia-induced PH, studies have shown that a significant amount of distal arteriole SMCs were derived from pre-existing smooth muscle cells [34]. It could be possible that these SMCs arising from Fgf10-positive cells during early development, could be one of the key cell types responsible for the increased muscularization of distal arterioles during hypoxia-induced PH. However, this needs further confirmation.
In a previous but nevertheless related study, Mlc1v-nLacZ-24transgenic mice (thereafter called Fgf10LacZ), in which the expression of LacZ was controlled by FGF10 regulatory sequences, were used to monitor the localization of Fgf10 positive cells. By using whole-mount in-situ hybridization and X-gal staining on the E12.5 and E13.5 embryonic lungs, it was found that β-galactosidase-positive cells were present exclusively in the distal mesenchyme, as well as adjacent to the bronchial epithelium. In addition, these β-galactosidase-positive cells around the bronchial epithelium also expressed the smooth muscle cell marker α-SMA suggesting that in the distal mesenchyme, the β-galactosidase-positive cells (Fgf10-positive cells) were progenitors for ASMCs. Reciprocal xenotransplantation on wild type and Fgf10LacZ/+embryos of the distal mesenchyme, as well as live imaging, demonstrated that β-galactosidase-positive cells in the distal mesenchyme were passively relocated around the bronchi [113].
In further studies, it was demonstrated that the Mlc1v-nLacZ-24allele behaved as an hypomorphic Fgf10 allele opening the way to generate allelic series of mice displaying different levels of FGF10 expression. These allelic series were generated by crossing the Fgf10LacZ transgenic line with Fgf10+/– mice on a C57Bl/6 background. Controls (Fgf10+/+) and Fgf10LacZ/– embryos were generated at different developmental stages.
By performing whole-mount immunohistochemistry with α-SMA antibodies in control and Fgf10LacZ/-, theauthorsfound that the reduction in Fgf10 expression led to decreased SMCs around bronchi, as well as a completely disorganized vasculature correlating with decreased VEGF expression [121].
Besides Fgf10 signaling, other FGFs are also involved in the formation of the pulmonary vasculature. White et al. demonstrated that both fibroblast growth factor 9 (Fgf9) and sonic hedgehog (SHH) signaling to the pulmonary mesenchyme (not the endothelial cells) was necessary and sufficient for the development of distal capillaries. However, Fgf9 could only partially reverse the decreased capillary density found without SHH signaling. SHH could not rescue the vascular phenotype of Fgf9null lungs. These results suggest that Fgf9 and SHH signaling synergistically regulate the growth and pattern of the pulmonary capillary plexus and regulate the temporal and spatial expression of Vegfa[54].FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network.
4. Conclusions
In conclusion, BPD-PH is affecting a population of preterm infants which should not be neglected, with considerably high burdens for the patients and the health care system. On the basis of data mainly gained from preclinical research in mice, it has become increasingly evident that vascular remodeling (VR) is the pathological basis for pulmonary hypertension in BPD.
However, the origin of the newly formed VSMCs during VR (in both the proximal arteries and the more distal arterioles) and their fate during reverse remodeling is not yet understood. In the future, lineage tracing experiments in the context of experimental BPD in mice (using the Sugen inhibitor 5416 to block VEGF signaling) will be instrumental to characterize the progenitors for these newly formed VSMCs in the disease context. Targeting these progenitor cells could provide an opportunity to treat the disease in human patients.
The BPD mouse model is a well-established and widely accepted model to mimicthe lung phenotype of BPD (alveolar simplification and dysmorphic lung vasculature) but in the context of BPD-PH, it needs to be mentioned that the only use of hyperoxia to induce lung injury is not causing PH in mice. A BPD-PH animal model needs to be established. It has been shown recently, that blockade of FGFR2b ligands activity postnatally in a BPD mouse model lead to decreased blood vessel number and increased muscularization resembling a PH-like phenotype [79]. Unpublished data also suggests that mice with constitutive FGF10/FGFR2b signaling deficiency (Fgf10+/-, Fgfr2b+/-) in room air are more susceptible to develop PH during aging as compared with wildtype mice.
FGF10 and FGFR2b signaling is believed to be essential in lung development. More interestingly, growing evidence coming from experimental research is confirming its preventive and regenerative effect during regeneration in lung diseases, such as BPD. In line with this notion, clinical-grade recombinant FGF10 could have therapeutic potential for the treatment of BPD-PH. However, based on the facts that other FGFs (e.g., FGF1 and FGF2) have been shown to be involved in the remodeling process of bronchial airways in COPD, thus, FGFs are potentially detrimental. Therefore, the therapeutic use of FGF10 needs to be thoroughly validated [134]. In addition, potential adverse side effects need to be evaluated [135]. As a first translational step, the authors suggest the validation of the preventive and therapeutic effect of recombinant FGF10 in a mouse model of BPD-PH. Large animal models would allow the intratracheal or inhalative administration of recombinant FGF10 which would be much more favorable and clinically relevant.
Apart from the abovementioned translational approaches, FGF10 expression (gene and protein levels) needs to be better clinically characterized within the heterogeneous cohort of preterm infants at risk of developing BPD-PH (e.g., gestation age, risk factors for BPD-PH, postnatal ventilation regimes, and genetic predisposition). Thus, future studies including the measurement, for example, using tracheal aspirates or exhaled breath condensate (EBC), of the expression of FGF10 in preterm infants is highly desirable. The establishment of a centralized biobank and databank would promote collection and characterization of biomaterials, provide organized availability and facilitate accessibility for research. In parallel to efforts invested in the clinical setting, more basic research needs to be done in order to gain deeper insight into the cellular and molecular mechanisms of FGF signaling governing vascular remodeling and reverse remodeling processes in BPD-PH.
The combination of both clinical and basic research would allow a better design of therapeutic strategies to prevent and to treat BPD-PH in the future.
This entry is adapted from the peer-reviewed paper 10.3390/cells9081875