Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Seong An | + 2110 word(s) | 2110 | 2021-12-03 03:55:47 | | | |
| 2 | Catherine Yang | Meta information modification | 2110 | 2021-12-03 06:38:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
An, S. Transthyretin. Encyclopedia. Available online: https://encyclopedia.pub/entry/16708 (accessed on 23 July 2026).
An S. Transthyretin. Encyclopedia. Available at: https://encyclopedia.pub/entry/16708. Accessed July 23, 2026.
An, Seong. "Transthyretin" Encyclopedia, https://encyclopedia.pub/entry/16708 (accessed July 23, 2026).
An, S. (2021, December 03). Transthyretin. In Encyclopedia. https://encyclopedia.pub/entry/16708
An, Seong. "Transthyretin." Encyclopedia. Web. 03 December, 2021.
Copy Citation
Transthyretin (TTR) is a thyroid hormone-binding protein which transports thyroxine from the bloodstream to the brain. The structural stability of TTR in tetrameric form is crucial for maintaining its original functions in blood or cerebrospinal fluid (CSF). The altered structure of TTR due to genetic mutations or its deposits due to aggregation could cause several deadly diseases such as cardiomyopathy and neuropathy in autonomic, motor, and sensory systems.
transthyretin
mutation
1. Introduction
Transthyretin (TTR) protein was discovered incidentally from cerebrospinal fluid (CSF) in 1942 and was called prealbumin based on its observed electrophoretic pattern [1]. The name “transthyretin” was formulated from the combination of three words: transport, thyroxine and retinol. As the name implied, TTR is a carrier protein of thyroxine and retinol. The gene for TTR is located in chromosome 18 position 12.1 (18q11.2-12.1) or base pair 31,591,766 to 31,599,023 and consists of four exons and five introns [2].
2. Structural Overview: The Role of TTR
TTR has a molecular weight of 14 kDa in monomeric form and exists in tetrameric form (55 kDa) in plasma [3]. It is synthesized in the liver and choroid plexus as a homo-tetramer structure with a dimer of dimers quaternary structure. The amino acid sequence from 1 to 20 is the signal peptide, and the final protein structure is expressed from amino acids 21 to 147. Hence, each monomer consists of 127 amino acids with one alpha helix, called EF helix, and eight beta strands (Figure 1a) [4][5]. Hydrogen bonds primarily kept the monomer and dimer structures intact. The pairs of hydrogen bonds in D strands of the monomers contributed to structural stability by forming an internal convex-shaped channel (Figure 1b) [6]. The two dimers then gathered to form a homo-tetramer through additional hydrogen bonds, finalizing the assembly of the carrier protein [7].
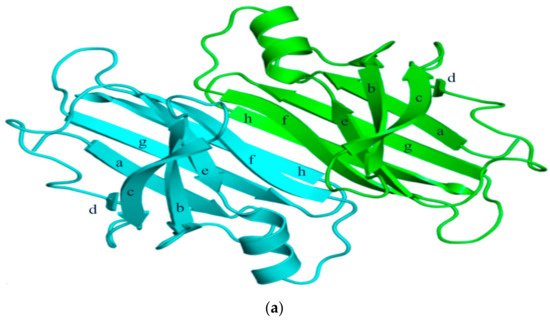
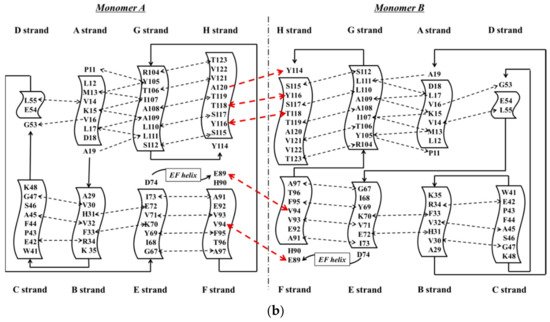
Figure 1. (a) 3-Dimensional structure of ATTRwt (PDBe ID code; 1BMZ [8]) in dimeric form; (b) Schematic diagram of hydrogen bonds in transthyretin and monomer–monomer’s hydrogen bonds. Arrowhead indicates acceptor, the tail indicates donor. In acidic condition, hydrogen bonds will be broken and destabilized.
Production of TTR by the liver or choroid plexus is followed by the translocation of the protein into the blood or CSF. The role of homo-tetrameric TTR is to transport T4 hormone and retinol. More than 99% of human T4 hormones are transported by carrier proteins, such as albumin, thyroxine-binding globulin, and TTR, and these same proteins are responsible for 15% of T4 hormone transports in circulation [9]. Homo-tetrameric TTR has two binding sites at a central channel called T4 pocket and is formed by side chain interactions [6]. Earlier reports of T4 binding studies predicted only one binding site, but investigation using a fluorescent probe, 8-anilinonapthalene-1-sulfonate, revealed two binding sites at opposite ends. Despite TTR having two T4 hormone-binding sites, the T4 hormone would preferentially bind to only one of the two sites. This preferential binding is influenced by relatively stronger binding affinity of one binding site and negative cooperativity, as well as structural changes of TTR, which can significantly reduce affinity at the other binding site [10]. Finally, the formation of hydrogen bonds between T4 hormone and Lys15 at the A strand/Glu54 at the D strand reinforced the hormone at the center of the tetramer (Figure 2) [11].
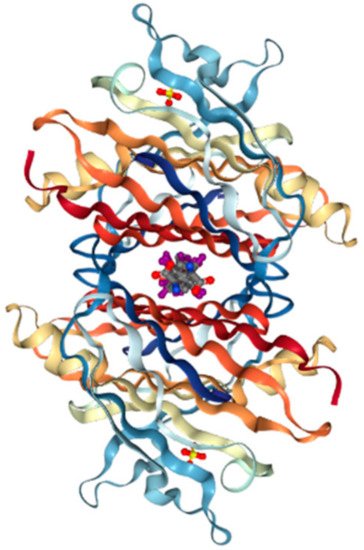
Figure 2. Transthyretin—T4 hormone binding structure. (PDB ID code; 2ROX [11]).
On the other hand, vitamin A (retinol) binds to TTR through a mediating protein, called retinol-binding protein (RBP) instead of the T4 binding protein. RBP (20 kDa) is synthesized by hepatocytes in the liver and consists of one alpha helix and eight anti-parallel beta-barrels [12]. Two RBPs bind and surround the TTR tetramer from both sides, but the limited concentration of RBP would result in a 1:1 molar ratio in plasma [13]. The amino acids of TTR interact with the following amino acids of RBP: Leu35, Trp67, Lys89, Trp91, Ser95, Phe96, Leu97 and Lys99 [14]. Retinol could not escape renal filtration through the urine once bound to the complex, TTR-RBP, thereby preserving retinol concentration as well as that of RBP [15][16]. The stability of the TTR-RBP-retinol complex is reduced when retinol is removed [17].
3. ATTR Causative Diseases
Mutations which are present in the TTR core structure reduce the stability of the protein. As a result, the TTR tetramer could separate into dimeric and monomeric forms, and these monomers coalesce by forming amyloid TTR (ATTR) aggregates (Figure 3) [18]. This aggregation, called amyloidogenesis, progresses to plaque formation, later depositing into tissues and neurons and inducing cytotoxicity. The different amyloidogenic diseases that present as a result were initially termed as the following: senile systemic amyloidosis (SSA), familial amyloid cardiomyopathy (FAC), and familial amyloid polyneuropathy (FAP) [18][19]. SSA, FAC, and FAP were reported to share the same accumulation of ATTR, but the types of ATTR found were different for each disease. In the case of SSA, aggregated constituents were composed mainly of large quantities of deposited wild-type ATTR (ATTRwt) in myocardia [20][21]. Updates in nomenclature were recently recommended by the International Society of Amyloidosis (ISA) to modify the terms SSA, FAC, and FAP to more exact definitions due to overlap in clinical presentation [22]. SSA was renamed as wild-type amyloid TTR amyloidosis (ATTRwt amyloidosis). It usually occurs in 25% of the elderly population over 80 with higher frequency in males at 25~50: 1 ratio [23][24]. Large amyloid deposits in the myocardia could cause congestive heart failure [25] and ventricular hypertrophy when echocardiography showed thickened ventricular walls along with low QRS voltages from electrocardiography (ECG) [26]. Common observable symptoms are fatigue, edema, shortness of breath, chest pain, and angina, and the severity of symptoms from these diseases may vary depending on the degree of ATTRwt deposits [27][28][29].
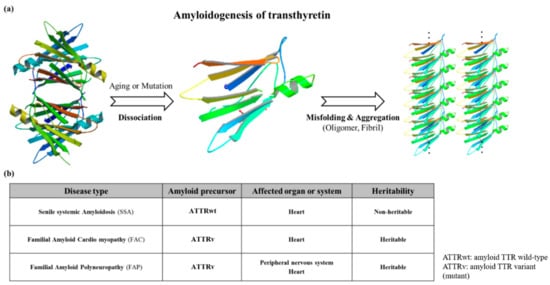
Figure 3. (a) Schematic diagram showing the amyloidogenesis of transthyretin. Transthyretin amyloidosis requires tetramer dissociation to monomer. (Transthyretin tetramer—PDB ID code; 4PVL83, Transthyretin monomer—PDB ID code; 3I9A84, Fibrils structure is made by ATTRv monomer without calculation); (b) Types of transthyretin amyloidoses.
FAC/FAP belong to the same pathological spectrum and differ from ATTRwt amyloidosis mainly due to ATTR mutations. In this case, ATTRv (amyloid TTR variant) refers to the mutant form of ATTR, and FAC/FAP was renamed to ATTR appended with the specific mutation that caused it (i.e., ATTRV30M) and the accompanying symptom (i.e. ATTR with cardiomyopathy) [22]. Depending on the mutations, the hydrophobic interaction of dimer–dimer formation would vary, resulting in the instability and breakdown of the tetramers. As a result, the tendency to produce the amyloid form is enhanced and favors the development of the disease [30]. FAP was formerly categorized into four types: FAP-I, FAP-II, FAP-III and FAP-IV, and the pathogenicity of FAP and FAC could overlap [31][32][33]. FAP-I has since been renamed as ATTRV30M amyloidosis and is also known as the Portuguese–Swedish–Japanese type, and haplotype comparison among foci suggested common Portuguese origins among Japanese, Spanish, and Brazilian patients, while Swedish patients had entirely different haplotypic origins [34][35]. ATTRV30M amyloidosis resulted in a wide range of symptoms such as sensory-autonomic, gastrointestinal and cardiac disturbances, impotence, and cardiomyopathy; as well as renal insufficiency in later stages of the disease [36][37]. A study on 15 Swedish families with ATTRV30M determined through Western blot showed the type of amyloid fibril composition remained the same despite the differences of the disease age of onset. Only one family showed different types of amyloid fibril composition on two brothers with similar ages of disease onset [38]. Despite the endemicity of the early onset form of ATTRV30M in some Portuguese, Japanese, and Swedish populations, the late onset form was shown to be more prevalent in non-endemic areas of Japan and Cyprus than previously thought, and much is still not known about its causative factors [39][40]. Symptoms of ATTRI84S amyloidosis were similar to ATTRV30M amyloidosis, but additional sensorimotor polyneuropathy could appear late in the disease [41]. In addition, amyloid deposits could be found in the eye, thyroid, adrenal glands, and blood vessels, leading to additional side effects from both diseases [42][43][44][45]. Even though ATTRv amyloidosis was like ATTRwt amyloidosis in terms of disease pathomechanism, the expression of ATTRv outside cardiac tissue may lead to amyloidosis resulting in hereditary ATTR cardiomyopathy (ATTR-CM). This was described in a Danish family carrying ATTRMet111 expressed in plasma that led to hereditary ATTR-CM (previously known as familial amyloid cardiomyopathy or FAC), whereas unafflicted members were seronegative [46]. Patients beyond the age of 60 were reported to have hereditary ATTR-CM, and ATTRV122I and ATTRT60A could be responsible. ATTRV122I was most commonly found in 4% of African-Americans, followed by ATTRT60A in patients from United Kingdom and Ireland [47][48][49]. The role of the non-coding variation of ATTRV122I was assessed in 4,361 unrelated African-Americans. The study showed that this allele increased 6.8-fold the risk of having ten or more outpatient surgeries. Additionally, men had a 15.2-fold higher risk of having ten or more outpatient surgeries. This non-coding variation seemed to accelerate the negative consequences associated with ATTRV122I amyloidosis [50]. Amyloid deposits in cardiac tissues may cause a thickening of cardiac walls leading to congestive heart failure and atrial arrhythmias, and result in cardiac arrest and death [45][51]. Carpal tunnel syndrome may manifest in the early phase of ATTRv amyloidosis and may require a great deal of attention and not simply dismissed as an unrelated symptom [52]. More than 140 ATTRv mutations have been identified [53] along with non-amyloidogenic mutations (Figure 4), which may not produce amyloid, but were reported to cause functional abnormalities in vivo [54][55]. Among non-amyloidogenic mutations, few were revealed to have a high affinity for thyroxine [54], and rarely, alanine mutations, particularly A109T and A109V, which were described in a family with dominantly-inherited euthyroid hyperthyroxinemia [56].

Figure 4. Transthyretin mutation sites and relation to amyloidosis.
4. General Diagnostic Workflow for ATTR Causative Diseases
Upon initial detection of the clinical symptoms mentioned above, additional diagnoses are still required to discern ATTR disease types for prompt treatment. ECG, magnetic resonance imaging (MRI), echocardiography, tissue biopsy, and genetic analysis could be performed for wild-type and hereditary ATTR amyloidoses. ECG is prioritized if the detected symptoms are primarily observed in the heart. In recent years, many kinds of ECG analysis were reported to lead to better initial diagnosis. As an example, left bundle branch block can differentiate ATTRwt amyloidosis from primary light chain amyloidosis (AL) as this ECG pattern can be observed in 40% of ATTRwt patients but is rare (4%) in AL [57]. Notably, low QRS voltages were observed in 60% of AL patients but were not as frequent in ATTRwt patients (40%) [57][58]. An abnormal ECG result would call for MRI using gadolinium enhancement by visualizing amyloid deposition in cardiac tissue for a more accurate diagnosis [59]. Echocardiography was used for diagnosing hypoplasia in the left and right ventricles of neonates [60] and has also been considered as an invaluable tool in providing real time and rapid evaluation of ventricular function in neonates and children with suspected ventricular anomalies [61]. This is largely in part due to its noninvasive nature and absence of side effects during right ventricular evaluation [62] which consists of assessment of ventricular atrophy based on ventricular muscle thickness [63]. Echocardiography also proved to be equally valuable in the assessment of left ventricular function through the measurement of different parametric velocities of the ventricular wall [64]. More recently, global longitudinal strain (GLS) measurement through speckle-tracking analysis of 2D-echocardiography was demonstrated to be a feasible non-invasive and more accurate alternative to the more traditional left ventricular ejection fraction (LVEF) [65]. Tissue biopsy could more accurately differentiate whether the disease was due to wild-type or variant-type ATTR deposits compared to simple visualization. If ATTR amyloidosis was suspected following MRI or 2D-echocardiography, tissue should be collected from the appropriate site depending on the patient’s condition, such as the heart tissue and sural nerve [66]. Each tissue was analyzed for ATTR types through amyloid fibril analysis using Congo red, immunohistochemistry, and mass spectrometry [67][68][69]. Despite its accuracy, acquiring tissue samples internally through biopsy is an invasive procedure when compared to ECG and 2D-echo, thus other more easily accessible organ sources were considered. Not too recently, skin biopsy was shown to be a promising alternative to the otherwise more invasive method of acquiring internal tissues from the heart or sural nerve in ATTRv (FAP) diagnosis [70]. Intraepidermal, sweat gland, and pilomotor nerve fiber densities were measured and compared in ATTRv patients, asymptomatic ATTRv carriers, healthy controls, diabetic neuropathy disease controls, and AL patients [70]. Immunohistochemistry revealed decreased fiber densities from all three tissue sources in ATTRv patients compared to normal controls, while ATTRv carriers showed intermediate reductions. The sensitivity and specificity for ATTRv diagnosis through detecting amyloid in skin was calculated to be 70% and 100%, respectively [70]. Additional genetic analyses using blood can be done to determine the type of disease. ATTR variants were identified by PCR-based full sequence analysis with 99% accuracy [71][72]. From these methods, patients with ATTRV112I were diagnosed as ATTR-CM (FAC at the time) [49], and the ATTRV30M variant could suggest amyloid deposition in the cardiac muscle or nerve [73]. If no mutation was found, the patient would be diagnosed with ATTRwt amyloidosis [20].
References
- Kabat, E.A.; Moore, D.H.; Landow, H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J. Clin. Investig. 1942, 21, 571.
- Wallace, M.R.; Naylor, S.L.; Kluve-Beckerman, B.; Long, G.L.; McDonald, L.; Shows, T.B.; Benson, M.D. Localization of the human prealbumin gene to chromosome 18. Biochem. Biophys. Res. Commun. 1985, 129, 753–758.
- Marchi, N.; Fazio, V.; Cucullo, L.; Kight, K.; Masaryk, T.; Barnett, G.; Volgelbaum, M.; Kinter, M.; Rasmussen, P.; Mayberg, M.R. Serum transthyretin monomer as a possible marker of blood-to-CSF barrier disruption. J. Neurosci. 2003, 23, 1949–1955.
- Xue, Q.; Zheng, Q.-C.; Zhang, J.-L.; Cui, Y.-L.; Chu, W.-T.; Zhang, H.-X. Mutation and low pH effect on the stability as well as unfolding kinetics of transthyretin dimer. Biophys. Chem. 2014, 189, 8–15.
- Yokoyama, T.; Mizuguchi, M.; Nabeshima, Y.; Kusaka, K.; Yamada, T.; Hosoya, T.; Ohhara, T.; Kurihara, K.; Tomoyori, K.; Tanaka, I. Hydrogen-bond network and pH sensitivity in transthyretin: Neutron crystal structure of human transthyretin. J. Struct. Biol. 2012, 177, 283–290.
- Blake, C.; Geisow, M.; Oatley, S.; Rerat, B.; Rerat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol. 1978, 121, 339–356.
- Prapunpoj, P.; Leelawatwattana, L. Evolutionary changes to transthyretin: Structure–function relationships. Febs J. 2009, 276, 5330–5341.
- Peterson, S.A.; Klabunde, T.; Lashuel, H.A.; Purkey, H.; Sacchettini, J.C.; Kelly, J.W. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1998, 95, 12956–12960.
- Liz, M.A.; Mar, F.M.; Franquinho, F.; Sousa, M.M. Aboard transthyretin: From transport to cleavage. IUBMB Life 2010, 62, 429–435.
- Ferguson, R.N.; Edelhoch, H.; Saroff, H.A.; Robbins, J.; Cahnmann, H.J. Negative cooperativity in the binding of thyroxine to human serum prealbumin. Biochemistry 1975, 14, 282–289.
- Wojtczak, A.; Cody, V.; Luft, J.R.; Pangborn, W. Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-N-acetyl-L-thyronine at 2.2 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 758–765.
- Melhus, H.; Bavik, C.-O.; Rask, L.; Peterson, P.A.; Eriksson, U. Epitope mapping of a monoclonal antibody that blocks the binding of retinol-binding protein to its receptor. Biochem. Biophys. Res. Commun. 1995, 210, 105–112.
- Newcomer, M.E.; Ong, D.E. Retinol Binding Protein and Its Interaction with Transthyretin; Landes Bioscience: Austin, TX, USA, 2000.
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041.
- Epstein, F.H.; Goodman, D.S. Vitamin A and retinoids in health and disease. New Engl. J. Med. 1984, 310, 1023–1031.
- Wolf, G. Multiple functions of vitamin A. Physiol. Rev. 1984, 64, 873–937.
- Noy, N.; Slosberg, E.; Scarlata, S. Interactions of retinol with binding proteins: Studies with retinol-binding protein and with transthyretin. Biochemistry 1992, 31, 11118–11124.
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.M.; Brito, R.M.M. Tetramer Dissociation and Monomer Partial Unfolding Precedes Protofibril Formation in Amyloidogenic Transthyretin Variants. J. Biol. Chem. 2001, 276, 27207–27213.
- Ruberg, F.L.; Judge, D.P.; Maurer, M.S. Familial Amyloid Cardiomyopathy Due to TTR Mutations: An underground Cause of Restrictive Cardiomyopathy. J. Card. Fail. 2009, 15, 464.
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845.
- Cornwell, G.G.; Sletten, K.; Johansson, B.; Westermark, P. Evidence that the amyloid fibril protein in senile systemic amyloidosis is derived from normal prealbumin. Biochem. Biophys. Res. Commun. 1988, 154, 648–653.
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219.
- Connors, S. Senile systemic amyloidosis presenting with heart failure. Arch. Intern. Med. 2005, 165, 1425–1429.
- Mohty, D.; Damy, T.; Cosnay, P.; Echahidi, N.; Casset-Senon, D.; Virot, P.; Jaccard, A. Cardiac amyloidosis: Updates in diagnosis and management. Arch. Cardiovasc. Dis. 2013, 106, 528–540.
- Hassan, W.; Al-Sergani, H.; Mourad, W.; Tabbaa, R. Amyloid heart disease: New frontiers and insights in pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2005, 32, 178.
- Dubrey, S.W.; Cha, K.; Simms, R.W.; Skinner, M.; Falk, R.H. Electrocardiography and Doppler echocardiography in secondary (AA) amyloidosis. Am. J. Cardiol. 1996, 77, 313–315.
- Drexler, M., Helmut; Coats, M.; Andrew, J.S. Explaining fatigue in congestive heart failure. Annu. Rev. Med. 1996, 47, 241–256.
- McKee, P.A.; Castelli, W.P.; McNamara, P.M.; Kannel, W.B. The natural history of congestive heart failure: The Framingham study. New Engl. J. Med. 1971, 285, 1441–1446.
- Ng, B.; Connors, L.H.; Davidoff, R.; Skinner, M.; Falk, R.H. Senile systemic amyloidosis presenting with heart failure: A comparison with light chain–associated amyloidosis. Arch. Intern. Med. 2005, 165, 1425–1429.
- Redondo, C.; Damas, A.M.; Olofsson, A.; Lundgren, E.; Saraiva, M.J.M. Search for intermediate structures in transthyretin fibrillogenesis: Soluble tetrameric Tyr78Phe TTR expresses a specific epitope present only in amyloid fibrils. J. Mol. Biol. 2000, 304, 461–470.
- Andrade, C. A peculiar form of peripheral neuropathy. Brain 1952, 75, 408–427.
- Akiya, S.; Nishio, Y.; Ibi, K.; Uozumi, H.; Takahashi, H.; Hamada, T.; Onishi, A.; Ishiguchi, H.; Hoshii, Y.; Nakazato, M. Lattice corneal dystrophy type II associated with familial amyloid polyneuropathy type IV. Ophthalmology 1996, 103, 1106–1110.
- Steiner, R.D.; Evans, J.P.; Paunio, T.; Uemichi, T.; Benson, M.D. Asp187Asn mutation of gelsolin in an American kindred with familial amyloidosis, Finnish type (FAP IV). Hum. Genet. 1995, 95, 327–330.
- Ohmori, H.; Ando, Y.; Makita, Y.; Onouchi, Y.; Nakajima, T.; Saraiva, M.; Terazaki, H.; Suhr, O.; Sobue, G.; Nakamura, M. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J. Med Genet. 2004, 41, e51.
- Zaros, C.; Genin, E.; Hellman, U.; Saporta, M.; Languille, L.; Wadington-Cruz, M.; Suhr, O.; Misrahi, M.; Planté-Bordeneuve, V. On the origin of the transthyretin Val30Met familial amyloid polyneuropathy. Ann. Hum. Genet. 2008, 72, 478–484.
- Hund, E.; Linke, R.; Willig, F.; Grau, A. Transthyretin-associated neuropathic amyloidosis pathogenesis and treatment. Neurology 2001, 56, 431–435.
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062.
- Suhr, O.B.; Wixner, J.; Anan, I.; Lundgren, H.-E.; Wijayatunga, P.; Westermark, P.; Ihse, E. Amyloid fibril composition within hereditary Val30Met (p. Val50Met) transthyretin amyloidosis families. PLoS ONE 2019, 14, e0211983.
- Koike, H.; Misu, K.-I.; Ikeda, S.-I.; Ando, Y.; Nakazato, M.; Ando, E.; Yamamoto, M.; Hattori, N.; Sobue, G.; Japan, F.T.S.G.F.H.N.I. Type I (Transthyretin Met30) Familial Amyloid Polyneuropathy in Japan: Early- vs. Late-Onset Form. JAMA Neurol. 2002, 59, 1771–1776.
- Andreou, S.; Panayiotou, E.; Michailidou, K.; Pirpa, P.; Hadjisavvas, A.; El Salloukh, A.; Barnes, D.; Antoniou, A.; Agathangelou, P.; Papastavrou, K.; et al. Epidemiology of ATTRV30M neuropathy in Cyprus and the modifier effect of complement C1q on the age of disease onset. Amyloid 2018, 25, 220–226.
- Sekijima, Y.; Yoshida, K.; Tokuda, T.; Ikeda, S.-I. Familial Transthyretin Amyloidosis; U.S. Department of Health & Human Services: Washington, DC, USA, 2018. Available online: https://rarediseases.info.nih.gov/diseases/656/familial-transthyretin-amyloidosis.
- Koga, T.; Ando, E.; Hirata, A.; Fukushima, M.; Kimura, A.; Ando, Y.; Negi, A.; Tanihara, H. Vitreous opacities and outcome of vitreous surgery in patients with familial amyloidotic polyneuropathy. Am. J. Ophthalmol. 2003, 135, 188–193.
- Misu, K.-i.; Hattori, N.; Nagamatsu, M.; Ikeda, S.-I.; Ando, Y.; Nakazato, M.; Takei, Y.-i.; Hanyu, N.; Usui, Y.; Tanaka, F. Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Brain 1999, 122, 1951–1962.
- OLOFSSON, B.O.; Grankvist, K.; Boman, K.; Forsberg, K.; Lafvas, I.; Lithner, F. Assessment of thyroid and adrenal function in patients with familial amyloidotic polyneuropathy. J. Intern. Med. 1989, 225, 337–341.
- Connection, B.H. Contemporary Reviews in Cardiovascular Medicine. Circulation 2007, 116, 77–84.
- Ranløv, I.; Alves, I.L.; Ranløv, P.J.; Husby, G.; Costa, P.P.; Saraiva, M.J. A Danish kindred with familial amyloid cardiomyopathy revisited: Identification of a mutant transthyretinmethionine 111 variant in serum from patients and carriers. Am. J. Med. 1992, 93, 3–8.
- Jacobson, D.R.; Pastore, R.D.; Yaghoubian, R.; Kane, I.; Gallo, G.; Buck, F.S.; Buxbaum, J.N. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. New Engl. J. Med. 1997, 336, 466–473.
- Sattianayagam, P.T.; Hahn, A.F.; Whelan, C.J.; Gibbs, S.D.; Pinney, J.H.; Stangou, A.J.; Rowczenio, D.; Pflugfelder, P.W.; Fox, Z.; Lachmann, H.J. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur. Heart J. 2012, 33, 1120–1127.
- Ruberg, F.L.; Maurer, M.S.; Judge, D.P.; Zeldenrust, S.; Skinner, M.; Kim, A.Y.; Falk, R.H.; Cheung, K.N.; Patel, A.R.; Pano, A. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am. Heart J. 2012, 164, 222–228.e221.
- Polimanti, R.; Nuñez, Y.Z.; Gelernter, J. Increased Risk of Multiple Outpatient Surgeries in African-American Carriers of Transthyretin Val122Ile Mutation Is Modulated by Non-Coding Variants. J. Clin. Med. 2019, 8, 269.
- Buxbaum, J.; Jacobson, D.R.; Tagoe, C.; Alexander, A.; Kitzman, D.W.; Greenberg, B.; Thaneemit-Chen, S.; Lavori, P. Transthyretin V122I in African Americans with congestive heart failure. J. Am. Coll. Cardiol. 2006, 47, 1724–1725.
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.-I.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet. J. Rare Dis. 2013, 8, 31.
- Rowczenio, D.M.; Noor, I.; Gillmore, J.D.; Lachmann, H.J.; Whelan, C.; Hawkins, P.N.; Obici, L.; Westermark, P.; Grateau, G.; Wechalekar, A.D. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum. Mutat. 2014, 35, E2403–E2412.
- Saraiva, M.J.M. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum. Mutat. 2001, 17, 493–503.
- Jacobson, D.R.; Alves, I.L.; Saraiva, M.J.; Thibodeau, S.N.; Buxbaum, J.N. Transthyretin Ser 6 gene frequency in individuals without amyloidosis. Hum. Genet. 1995, 95, 308–312.
- Refetoff, S.; Marinov, V.; Tunca, H.; Byrne, M.M.; Sunthornthepvarakul, T.; Weiss, R. A new family with hyperthyroxinemia caused by transthyretin Val109 misdiagnosed as thyrotoxicosis and resistance to thyroid hormone--a clinical research center study. J. Clin. Endocrinol. Metab. 1996, 81, 3335–3340.
- Banypersad, S.M.; Moon, J.C.; Whelan, C.; Hawkins, P.N.; Wechalekar, A.D. Updates in cardiac amyloidosis: A review. J. Am. Heart Assoc. 2012, 1, e000364.
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 2009, 120, 1203–1212.
- Maceira, A.M.; Joshi, J.; Prasad, S.K.; Moon, J.C.; Perugini, E.; Harding, I.; Sheppard, M.N.; Poole-Wilson, P.A.; Hawkins, P.N.; Pennell, D.J. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005, 111, 186–193.
- MEYER, R.A.; KAPLAN, S. Echocardiography in the Diagnosis of Hypoplasia of the Left or Right Ventricles in the Neonate. Circulation 1972, 46, 55–64.
- Tissot, C.; Singh, Y.; Sekarski, N. Echocardiographic Evaluation of Ventricular Function—For the Neonatologist and Pediatric Intensivist. Front. Pediatrics 2018, 6.
- Jurcut, R.; Giusca, S.; La Gerche, A.; Vasile, S.; Ginghina, C.; Voigt, J.-U. The echocardiographic assessment of the right ventricle: What to do in 2010? Eur. Heart J. Cardiovasc. Imaging 2010, 11, 81–96.
- Choudhary, G.M.; Arushi, A.; Stapleton, D.; Reddy, P.C. Assessment of right ventricle by echocardiogram. In Echocardiography in Heart Failure and Cardiac Electrophysiology; Lakshmanadoss, U., Ed.; IntechOpen Limited: London, UK, 19 October 2016.
- Quinones, M.A.; Gaasch, W.H.; Alexander, J.K. Echocardiographic assessment of left ventricular function with special reference to normalized velocities. Circulation 1974, 50, 42–51.
- Potter, E.; Marwick, T.H. Assessment of Left Ventricular Function by Echocardiography: The Case for Routinely Adding Global Longitudinal Strain to Ejection Fraction. JACC: Cardiovasc. Imaging 2018, 11, 260–274.
- Sousa, M.M.; Cardoso, I.; Fernandes, R.; Guimaraes, A.; Saraiva, M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: Evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 2001, 159, 1993–2000.
- Ihse, E.; Ybo, A.; Suhr, O.B.; Lindqvist, P.; Backman, C.; Westermark, P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J. Pathol. 2008, 216, 253–261.
- Jamet, M.-P.; Gnemmi, V.; Hachulla, É.; Dhaenens, C.-M.; Bouchindhomme, B.; Delattre, C.; Glowacki, F.; Hatron, P.-Y.; Lacour, A.; Lamblin, N. Distinctive patterns of transthyretin amyloid in salivary tissue: A clinicopathologic study of 92 patients with amyloid-containing minor salivary gland biopsies. Am. J. Surg. Pathol. 2015, 39, 1035–1044.
- Satoskar, A.A.; Efebera, Y.; Hasan, A.; Brodsky, S.; Nadasdy, G.; Dogan, A.; Nadasdy, T. Strong tranthyretin immunostaining, potential pitfall in cardiac amyloid typing. Am. J. Surg. Pathol. 2011, 35, 1685.
- Ebenezer, G.J.; Liu, Y.; Judge, D.P.; Cunningham, K.; Truelove, S.; Carter, N.D.; Sebastian, B.; Byrnes, K.; Polydefkis, M. Cutaneous nerve biomarkers in transthyretin familial amyloid polyneuropathy. Ann. Neurol. 2017, 82, 44–56.
- Soares, M.L.; Coelho, T.; Sousa, A.; Batalov, S.; Conceição, I.; Sales-Luís, M.L.; Ritchie, M.D.; Williams, S.M.; Nievergelt, C.M.; Schork, N.J. Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: Complexity in a single-gene disease. Hum. Mol. Genet. 2005, 14, 543–553.
- Yamashita, T.; Asl, K.H.; Yazaki, M.; Benson, M.D. A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid 2005, 12, 127–130.
- Tojo, K.; Sekijima, Y.; Kelly, J.W.; Ikeda, S.-I. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci. Res. 2006, 56, 441–449.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
03 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No