Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kazunori Kageyama | + 1274 word(s) | 1274 | 2021-11-16 10:58:56 | | | |
| 2 | Yvaine Wei | Meta information modification | 1274 | 2021-11-26 08:37:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kageyama, K. Hypothalamic Regulation of Corticotropin-Releasing Factor under Stress. Encyclopedia. Available online: https://encyclopedia.pub/entry/16417 (accessed on 22 July 2026).
Kageyama K. Hypothalamic Regulation of Corticotropin-Releasing Factor under Stress. Encyclopedia. Available at: https://encyclopedia.pub/entry/16417. Accessed July 22, 2026.
Kageyama, Kazunori. "Hypothalamic Regulation of Corticotropin-Releasing Factor under Stress" Encyclopedia, https://encyclopedia.pub/entry/16417 (accessed July 22, 2026).
Kageyama, K. (2021, November 26). Hypothalamic Regulation of Corticotropin-Releasing Factor under Stress. In Encyclopedia. https://encyclopedia.pub/entry/16417
Kageyama, Kazunori. "Hypothalamic Regulation of Corticotropin-Releasing Factor under Stress." Encyclopedia. Web. 26 November, 2021.
Copy Citation
Stress response is considered the physiological and behavioral response to internal or external stimulus. Corticotropin-releasing factor (CRF) in the hypothalamus plays a central role in regulating the stress response. CRF stimulates adrenocorticotropic hormone (ACTH) release from the anterior pituitary. ACTH stimulates glucocorticoid secretion from the adrenal glands. Glucocorticoids are essential for stress coping, stress resilience, and homeostasis.
glucocorticoid
hypothalamus
corticotropin-releasing factor
stress
1. Introduction
The stress response is adaptive, and stress resilience is regarded as the result of the adaptive response to a stressor [1][2]. The hypothalamic-pituitary-adrenal (HPA) axis is activated under various stressors (Figure 1A). Limbic structures, such as the central amygdala, bed nuclei of the stria terminalis, and nucleus accumbens shell of the extended amygdala, and the hippocampus, play an important role in stress responses. In parallel, stress responses also activate a physiological system for stress adaptation (Figure 1B).
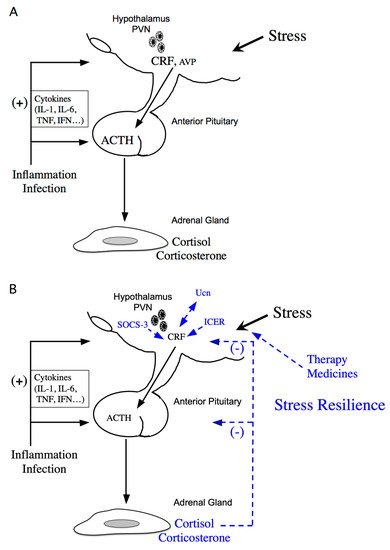
Figure 1. Schematic model of hypothalamic-pituitary-adrenal (HPA) axis regulation. (A) Activation of the HPA axis under stress. Corticotropin-releasing factor (CRF) plays a central role in controlling stress responses. Cytokines also stimulate CRF production under inflammation or infection. CRF, produced in the hypothalamic paraventricular nucleus (PVN), stimulates adrenocorticotropic hormone (ACTH) production from the corticotrophs of the anterior pituitary (AP). ACTH then stimulates corticosterone and cortisol release, the principal glucocorticoids in rodents and human, respectively, from the adrenal glands. (B) Regulation of the HPA axis under stress resilience. Glucocorticoids are produced by ACTH in the adrenal glands. Circulating glucocorticoids are critical for recovery from stress conditions. Both inducible cAMP-early repressor (ICER) and suppressor of cytokine signaling (SOCS-3) contribute to the negative regulation of CRF synthesis in the hypothalamus. The urocortin (Ucn)-CRF receptor type 2 mediates “stress coping” responses. Therapy or medicines also target this to alleviate stressed states.
2. Molecular Mechanisms of CRF Regulation in the Hypothalamus
Many neurotransmitters and neuropeptides are involved in activation of CRF neurons in the hypothalamus. Serotonin, noradrenaline, neuromedin C, and thyrotropin-releasing hormone affect intracellular Ca2+ concentration in CRF neurons of the PVN [3] and activate CRF neurons. Pituitary adenylate cyclase-activating polypeptide (PACAP), a member of the secretin/glucagon/vasoactive intestinal peptide family, and glucagon-like peptide 1 (GLP1) also stimulate Crf gene activity in hypothalamic cells [4][5]. PACAP can contribute to CRF activation in the hypothalamus under emotional stress. In fact, the extended amygdala and the bed nuclei of the stria terminalis are identified as innervation sites of PACAP neurons. Both PACAP and the PACAP-selective PACAP receptor type 1 are highly expressed in the hypothalamus and the supraoptic nucleus [6][7]. PACAP increases Crf mRNA levels in the parvocellular region of the PVN, suggesting its involvement in the positive regulation of Crf gene expression [8]. The cyclic AMP (cAMP)-protein kinase A (PKA) pathway is involved in CRF synthesis by PACAP [4]. GLP1 also stimulates the activities of both Crf and Avp promoters in hypothalamic cells [5]. Basal promoter activities of Crf and Avp are increased in high glucose medium [5], while Crf and Avp promoter activities are increased by GLP1 in standard or low glucose medium but not in high. Hyperglycemia is, therefore, a stressor increasing the synthesis of CRF and AVP in the hypothalamus.
Stress resilience is considered the result of the adaptive response to a stressor. Circulating glucocorticoids are critical for recovery from stress conditions and essential for stress resilience (Figure 1B). Treatment with glucocorticoids in adrenalectomized rats does not perfectly prevent the increase in stress-induced Crf heteronuclear RNA [9]. Therefore, factors other than glucocorticoids might be involved in limiting CRF activation during stress. Inducible cAMP-early repressor (ICER), a cAMP-inducible member of the CRE modulator (CREM) family and a CREM repressor isoform, are such candidates [10][11]. CREM, CREB, and AP-1 bind to CRE promoter elements to stimulate transcription [12], while ICER acts as a competitive inhibitor of such CRE-dependent transcription [10]. Therefore, ICER can suppress the stress response via inhibition of the cAMP-dependent Crf gene [13] (Figure 1B). Suppressor of cytokine signaling (SOCS)-3 acts as a potent negative regulator of cytokine signaling [14]. SOCS-3 suppresses cytokine-induced ACTH production in corticotrophs [15]. IL-6 stimulates Janus kinase/signal transducers and activators of the transcription (JAK/STAT) signaling, while IL-6-induced SOCS-3 acts as a negative regulator and inhibits STAT phosphorylation by JAK at the receptor complex [16][17]. SOCS-3 is stimulated by IL-6 and cAMP, while SOCS-3 knockdown increases IL-6- or forskolin-induced Crf gene transcription, thereby contributing to the negative regulation of CRF synthesis in the hypothalamus [18] (Figure 1B).
3. Roles of the CRF Peptide Family and Stress-Related Peptides and Their Receptors under Stress
Urocortins (Ucns) are members of the CRF peptide family. Three Ucns have been found in mammals. Ucn1 has potent effects including appetite suppression and modulation of the cardiovascular system [19][20][21]. Ucn2 has more potent vasodilatory and cardiac inotropic effects than CRF and shows suppression of host resistance to infection via IL-10 upregulation [22]. Ucn3 modulates insulin secretion and Ca2+ influx in pancreatic β-cells [23] and improves cellular stress responses and glucose uptake [24].
The actions of the CRF family peptides are mediated by ≥ 2 distinct G protein-coupled receptors, namely the CRF receptor type 1 (CRF1 receptor) [25][26][27] and CRF receptor type 2 (CRF2 receptor) [28][29][30]. CRF has a higher affinity for the CRF1 receptor than for the CRF2 receptor (Figure 2) [31]. Ucn1 binds to both the CRF1 and CRF2 receptors, while Ucn2 and Ucn3 are highly selective for the CRF2 receptor, with little affinity for the CRF1 receptor (Figure 2) [19][32][33][34].
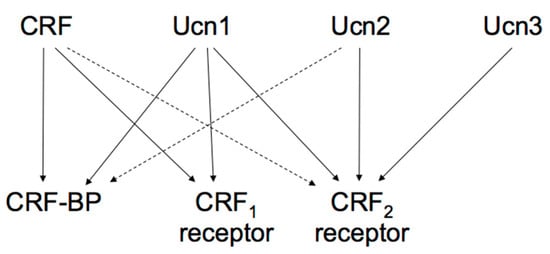
Figure 2. Proposed signaling mechanisms of corticotropin-releasing factor (CRF), urocortins (Ucns) and CRF receptors. CRF-BP, CRF-binding protein. Solid lines represent a high affinity binding, and dashed lines a low affinity binding.
These two receptors share 69% amino acid homology [29] but have different tissue distributions and pharmacological properties with respect to ligands [31]. CRF1 receptor is mainly expressed in the pituitary, the brain, and various peripheral tissues. In pituitary corticotrophs, CRF1 receptor is the major subtype responsible for regulating ACTH synthesis and secretion. CRF2 receptor is located in different brain areas than the CRF1 receptor and is also abundant in the periphery. The CRF2 receptor has ≥ 3 alternative splice variants, CRF2a receptor, CRF2b receptor, and CRF2g receptor. In the rat, CRF2a receptor mRNA was found primarily in the brain and the pituitary [35][36]. In contrast, the CRF2b receptor was predominantly expressed in peripheral sites such as the heart, gastrointestinal tract, and vascular smooth muscles [36]. CRF receptors primarily activate adenylyl cyclase and cAMP pathways through Gsα activation. In addition, CRF receptors can activate other types of Gα [37].
4. Negative CRF Feedback Mechanisms in the Hypothalamus
Glucocorticoids inhibit CRF production in the hypothalamic PVN and ACTH production in the AP, modulating its own production (Figure 1B). Simultaneously, glucocorticoids inhibit CRF neurons in the PVN by binding to GRs in the hippocampus [38].
The HPA axis is regulated by a negative feedback mechanism (Figure 1B). In fact, hypothalamic parvocellular neurons express GRs, and glucocorticoids negatively regulate Crf gene expression directly in the hypothalamus. The Crf promoter region contributes to inhibition by glucocorticoids by acting as a negative GRE (nGRE) (Figure 3), even though the Crf promoter does not contain a classical consensus GRE, there are several regions GRs can bind to [39][40]. Glucocorticoids can inhibit CREB and cFos in the PVN [41][42][43].

Figure 3. Schematic model of transcriptional regulation of the corticotropin-releasing factor (Crf) promoter. Possible binding sites for transcriptional factors such as the cAMP-response element (CRE), activator protein 1 (AP-1) protein (Fos/Jun) binding sites, the half glucocorticoid regulatory element (GRE), and the half estrogen-responsive element (ERE) in the proximal Crf promoter. The Crf promoter region also contributes to glucocorticoid inhibition as a negative GRE (nGRE), as it includes a serum response element (SRE).
Additionally, other promoter regions are involved in the inhibition of Crf gene expression in hypothalamic cells (Figure 3). Crf promoter sequences between –248 and –233 bp are also involved in the glucocorticoid suppression of cAMP-stimulated Crf promoter activity (Figure 3), including a serum response element (SRE) [44], which contributes to the negative response to glucocorticoids, because the GR can bind to SRE and inhibit promoter activation by antagonizing positive transcription [44]. Therefore, in addition to nGRE, the glucocorticoid suppression of cAMP stimulated CRF promoter activity may also be caused by SRE in hypothalamic cells.
References
- Oken, B.S.; Chamine, I.; Wakeland, W. A systems approach to stress, stressors and resilience in humans. Behav. Brain Res. 2015, 282, 144–154.
- Leon, M.A.G.; Pérez-Mármol, J.M.; Gonzalez-Pérez, R.; García-Ríos, M.D.C.; Peralta-Ramírez, M.I. Relationship between resilience and stress: Perceived stress, stressful life events, HPA axis response during a stressful task and hair cortisol. Physiol. Behav. 2019, 202, 87–93.
- Mukai, Y.; Nagayama, A.; Itoi, K.; Yamanaka, A. Identification of substances which regulate activity of corticotropin-releasing factor-producing neurons in the paraventricular nucleus of the hypothalamus. Sci. Rep. 2020, 10, 1–14.
- Kageyama, K.; Hanada, K.; Iwasaki, Y.; Sakihara, S.; Nigawara, T.; Kasckow, J.; Suda, T. Pituitary adenylate cyclase-activating polypeptide stimulates corticotropin-releasing factor, vasopressin and interleukin-6 gene transcription in hypothalamic 4B cells. J. Endocrinol. 2007, 195, 199–211.
- Kageyama, K.; Yamagata, S.; Akimoto, K.; Sugiyama, A.; Murasawa, S.; Suda, T. Action of glucagon-like peptide 1 and glucose levels on corticotropin-releasing factor and vasopressin gene expression in rat hypothalamic 4B cells. Mol. Cell. Endocrinol. 2012, 362, 221–226.
- Nomura, M.; Ueta, Y.; Serino, R.; Kabashima, N.; Shibuya, I.; Yamashita, H. PACAP type I receptor gene expression in the paraventricular and supraoptic nuclei of rats. NeuroReport 1996, 8, 67–70.
- Shioda, S.; Shuto, Y.; Somogyvári-Vigh, A.; Legradi, G.; Onda, H.; Coy, D.H.; Nakajo, S.; Arimura, A. Localization and gene expression of the receptor for pituitary adenylate cyclase-activating polypeptide in the rat brain. Neurosci. Res. 1997, 28, 345–354.
- Grinevich, V.; Fournier, A.; Pelletier, G. Effects of pituitary adenylate cyclase-activating polypeptide (PACAP) on corticotropin-releasing hormone (CRH) gene expression in the rat hypothalamic paraventricular nucleus. Brain Res. 1997, 773, 190–196.
- Ma, X.M.; Aguilera, G. Differential regulation of corticotropin-releasing hormone and vasopressin transcription by glucocorticoids. Endocrinology 1999, 140, 5642–5650.
- Foulkes, N.S.; Borrelli, E.; Sassone-Corsi, P. CREM gene: Use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell 1991, 64, 739–749.
- Molina, C.A.; Foulkes, N.S.; Lalli, E.; Sassone-Corsi, P. Inducibility and negative autoregulation of CREM: An alternative promoter directs the expression of ICER, an early response repressor. Cell 1993, 75, 875–886.
- Lalli, E.; Sassone-Corsi, P. Signal transduction and gene regulation: The nuclear response to cAMP. J. Biol. Chem. 1994, 269, 17359–17362.
- Kageyama, K.; Tamasawa, N.; Suda, T. Signal transduction in the hypothalamic corticotropin-releasing factor system and its clinical implications. Stress 2011, 14, 357–367.
- Krebs, D.; Hilton, D. SOCS: Physiological suppressors of cytokine signaling. J. Cell Sci. 2000, 113, 2813–2819.
- Auernhammer, C.J.; Bousquet, C.; Melmed, S. Autoregulation of pituitary corticotroph SOCS-3 expression: Characterization of the murine SOCS-3 promoter. Proc. Natl. Acad. Sci. USA 1999, 96, 6964–6969.
- Ram, P.A.; Waxman, D. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J. Biol. Chem. 1999, 274, 35553–35561.
- Schmitz, J.; Weissenbach, M.; Haan, S.; Heinrich, P.C.; Schaper, F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000, 275, 12848–12856.
- Kageyama, K.; Hanada, K.; Iwasaki, Y.; Suda, T. Regulation and role of suppressor of cytokine signaling-3 in hypothalamic 4B cells. J. Endocrinol. 2009, 201, 369–376.
- Vaughan, J.; Donaldson, C.J.; Bittencourt, J.; Perrin, M.H.; Lewis, A.K.; Sutton, S.; Chan, R.; Turnbull, A.V.; Lovejoy, D.; Rivier, C.; et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nat. Cell Biol. 1995, 378, 287–292.
- Spina, M.; Merlo-Pich, E.; Chan, R.K.W.; Basso, A.M.; Rivier, J.; Vale, W.; Koob, G.F. Appetite-suppressing effects of urocortin, a CRF-related neuropeptide. Science 1996, 273, 1561–1564.
- Parkes, D.G.; Vaughan, J.; Rivier, J.; Vale, W.; May, C.N. Cardiac inotropic actions of urocortin in conscious sheep. Am. J. Physiol. Content 1997, 272, H2115–H2122.
- Sashinami, H.; Kageyama, K.; Suda, T.; Nakane, A. Urocortin 2 suppresses host resistance to Listeria monocytogenes infection via up-regulation of interleukin-10. Endocrinology 2005, 146, 5003–5011.
- Kageyama, K.; Kimura, R.; Suga, S.; Ogawa, Y.; Suda, T.; Wakui, M. Modulation of Ca2+ influx by corticotropin-releasing factor (CRF) family of peptides via CRF receptors in rat pancreatic beta-cells. Peptides 2006, 27, 1814–1819.
- Kavalakatt, S.; Khadir, A.; Madhu, D.; Koistinen, H.A.; Al-Mulla, F.; Tuomilehto, J.; Abubaker, J.; Tiss, A. Urocortin 3 overexpression reduces ER stress and heat shock response in 3T3-L1 adipocytes. Sci. Rep. 2021, 11, 1–11.
- Chang, C.P.; Pearse, R.; O’Connell, S.; Rosenfeld, M.G. Identification of a seven transmembrane helix receptor for corticotropin-releasing factor and sauvagine in mammalian brain. Neuron 1993, 11, 1187–1195.
- Chen, R.; Lewis, K.A.; Perrin, M.H.; Vale, W.W. Expression cloning of a human corticotropin-releasing-factor receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 8967–8971.
- Vita, N.; Laurent, P.; Lefort, S.; Chalon, P.; Lelias, J.-M.; Kaghad, M.; Le Fur, G.; Caput, D.; Ferrara, P. Primary structure and functional expression of mouse pituitary and human brain corticotrophin releasing factor receptors. FEBS Lett. 1993, 335, 1–5.
- Lovenberg, T.W.; Liaw, C.W.; Grigoriadis, D.E.; Clevenger, W.; Chalmers, D.T.; De Souza, E.B.; Oltersdorf, T. Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc. Natl. Acad. Sci. USA 1995, 92, 836–840.
- Perrin, M.; Donaldson, C.; Chen, R.; Blount, A.; Berggren, T.; Bilezikjian, L.; Sawchenko, P.; Vale, W. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc. Natl. Acad. Sci. USA 1995, 92, 2969–2973.
- Stenzel, P.; Kesterson, R.; Yeung, W.; Cone, R.D.; Stenzel-Poore, M.P.; Rittenberg, M.B. Identification of a novel murine receptor for corticotropin-releasing hormone expressed in the heart. Mol. Endocrinol. 1995, 9, 637–645.
- Suda, T.; Kageyama, K.; Sakihara, S.; Nigawara, T. Physiological roles of urocortins, human homologues of fish urotensin I, and their receptors. Peptides 2004, 25, 1689–1701.
- Hsu, S.Y.; Hsueh, A.J. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat. Med. 2001, 7, 605–611.
- Lewis, K.; Li, C.; Perrin, M.H.; Blount, A.; Kunitake, K.; Donaldson, C.; Vaughan, J.; Reyes, T.M.; Gulyas, J.; Fischer, W.; et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7570–7575.
- Reyes, T.M.; Lewis, K.; Perrin, M.H.; Kunitake, K.S.; Vaughan, J.; Arias, C.A.; Hogenesch, J.B.; Gulyas, J.; Rivier, J.; Vale, W.W.; et al. Urocortin II: A member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 2843–2848.
- Kageyama, K.; Li, C.; Vale, W.W. Corticotropin-releasing factor receptor type 2 messenger ribonucleic acid in rat pituitary: Localization and regulation by immune challenge, restraint stress, and glucocorticoids. Endocrinology 2003, 144, 1524–1532.
- Lovenberg, T.W.; Chalmers, D.T.; Liu, C.; De Souza, E.B. CRF2 alpha and CRF2 beta receptor mRNAs are differentially distributed between the rat central nervous system and peripheral tissues. Endocrinology 1995, 136, 4139–4142.
- Vasconcelos, M.; Stein, D.J.; Gallas-Lopes, M.; Landau, L.; De Almeida, R.M.M. Corticotropin-releasing factor receptor signaling and modulation: Implications for stress response and resilience. Trends Psychiatry Psychother. 2020, 42, 195–206.
- Ding, H.; Cui, S.Y.; Cui, X.Y.; Liu, Y.T.; Hu, X.; Zhao, H.L.; Qin, Y.; Kurban, N.; Zhang, Y.H. Anti-stress effects of combined block of glucocorticoid and mineralocorticoid receptors in the paraventricular nucleus of the hypothalamus. Br. J. Pharmacol. 2021, 178, 3696–3707.
- Guardiola-Diaz, H.M.; Kolinske, J.S.; Gates, L.H.; Seasholtz, A.F. Negative glucorticoid regulation of cyclic adenosine 3’, 5’-monophosphate-stimulated corticotropin-releasing hormone-reporter expression in AtT-20 cells. Mol. Endocrinol. 1996, 10, 317–329.
- Malkoski, S.P.; Dorin, R.I. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol. Endocrinol. 1999, 13, 1629–1644.
- King, B.R.; Smith, R.; Nicholson, R.C. Novel glucocorticoid and cAMP interactions on the CRH gene promoter. Mol. Cell. Endocrinol. 2002, 194, 19–28.
- Jacobson, L.; Sharp, F.R.; Dallman, M.F. Induction of fos-like immunoreactivity in hypothalamic corticotropin-releasing factor neurons after adrenalectomy in the rat. Endocrinology 1990, 126, 1709–1719.
- Légrádi, G.; Holzer, D.; Kapcala, L.P.; Lechan, R.M. Glucocorticoids inhibit stress-induced phosphorylation of CREB in corticotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Neuroendocrinology 1997, 66, 86–97.
- Karagianni, N.; Tsawdaroglou, N. The c-fos serum response element (SRE) confers negative response to glucocorticoids. Oncogene 1994, 9, 2327–2334.
More
Information
Subjects:
Endocrinology & Metabolism
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Entry Collection:
Peptides for Health Benefits
Revisions:
2 times
(View History)
Update Date:
27 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No