 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Feroza K Choudhury | + 1902 word(s) | 1902 | 2021-11-23 05:00:20 | | | |
| 2 | Vivi Li | Meta information modification | 1902 | 2021-11-24 02:25:25 | | |
Video Upload Options
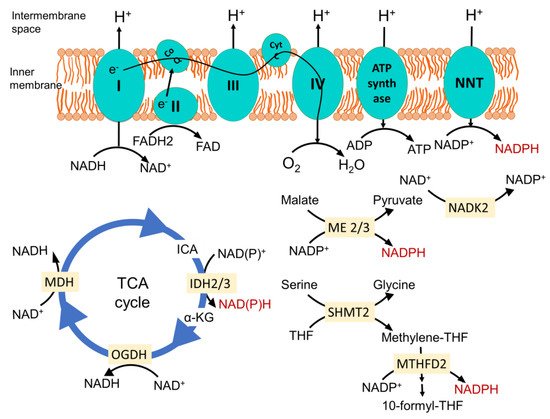
Mitochondrial redox metabolism is the central component in the cellular metabolic landscape, where anabolic and catabolic pathways are reprogrammed to maintain optimum redox homeostasis. During different stages of cancer, the mitochondrial redox status plays an active role in navigating cancer cells’ progression and regulating metabolic adaptation according to the constraints of each stage. Mitochondrial reactive oxygen species (ROS) accumulation induces malignant transformation. Once vigorous cell proliferation renders the core of the solid tumor hypoxic, the mitochondrial electron transport chain mediates ROS signaling for bringing about cellular adaptation to hypoxia. Highly aggressive cells are selected in this process, which are capable of progressing through the enhanced oxidative stress encountered during different stages of metastasis for distant colonization. Mitochondrial oxidative metabolism is suppressed to lower ROS generation, and the overall cellular metabolism is reprogrammed to maintain the optimum NADPH level in the mitochondria required for redox homeostasis. After reaching the distant organ, the intrinsic metabolic limitations of that organ dictate the success of colonization and flexibility of the mitochondrial metabolism of cancer cells plays a pivotal role in their adaptation to the new environment.
1. Introduction
2. Mitochondrial Redox Metabolism
References
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 4862.
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721.
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528.
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807.
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182.e4.
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176.
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253.
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98.
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473.
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676.
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17.
- Sullivan, L.B.; Gui, D.Y.; Vander Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693.
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752.
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338.
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569.
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325.
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637.
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390.
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997.
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218.
- Ismail, T.; Kim, Y.; Lee, H.; Lee, D.S.; Lee, H.S. Interplay between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression. Int. J. Mol. Sci. 2019, 20, 4407.
- Kampjut, D.; Sazanov, L.A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature 2019, 573, 291–295.
- Cracan, V.; Titov, D.V.; Shen, H.; Grabarek, Z.; Mootha, V.K. A genetically encoded tool for manipulation of NADP+/NADPH in living cells. Nat. Chem. Biol. 2017, 13, 1088–1095.
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263.
- Nesci, S.; Trombetti, F.; Pagliarani, A. Nicotinamide Nucleotide Transhydrogenase as a Sensor of Mitochondrial Biology. Trends Cell Biol. 2020, 30, 1–3.
- Rao, K.N.S.; Shen, X.; Pardue, S.; Krzywanski, D.M. Nicotinamide nucleotide transhydrogenase (NNT) regulates mitochondrial ROS and endothelial dysfunction in response to angiotensin II. Redox Biol. 2020, 36, 101650.
- Lee, J.-H.; Go, Y.; Kim, D.-Y.; Lee, S.H.; Kim, O.-H.; Jeon, Y.H.; Kwon, T.K.; Bae, J.-H.; Song, D.-K.; Rhyu, I.J.; et al. Isocitrate dehydrogenase 2 protects mice from high-fat diet-induced metabolic stress by limiting oxidative damage to the mitochondria from brown adipose tissue. Exp. Mol. Med. 2020, 52, 238–252.
- Hsieh, J.-Y.; Shih, W.-T.; Kuo, Y.-H.; Liu, G.-Y.; Hung, H.-C. Functional Roles of Metabolic Intermediates in Regulating the Human Mitochondrial NAD(P)+-Dependent Malic Enzyme. Sci. Rep. 2019, 9, 9081.
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302.
- Ohashi, K.; Kawai, S.; Murata, K. Identification and characterization of a human mitochondrial NAD kinase. Nat. Commun. 2012, 3, 1248.
- Tran, D.H.; Kesavan, R.; Rion, H.; Soflaee, M.H.; Solmonson, A.; Bezwada, D.; Vu, H.S.; Cai, F.; Phillips, J.A.; DeBerardinis, R.J.; et al. Mitochondrial NADP+ is essential for proline biosynthesis during cell growth. Nat. Metab. 2021, 3, 571–585.
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452.
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125.
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812.
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639.
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667.
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904.
- Hollinshead, K.E.R.; Munford, H.; Eales, K.L.; Bardella, C.; Li, C.; Escribano-Gonzalez, C.; Thakker, A.; Nonnenmacher, Y.; Kluckova, K.; Jeeves, M.; et al. Oncogenic IDH1 Mutations Promote Enhanced Proline Synthesis through PYCR1 to Support the Maintenance of Mitochondrial Redox Homeostasis. Cell Rep. 2018, 22, 3107–3114.
- Liu, W.; Glunde, K.; Bhujwalla, Z.M.; Raman, V.; Sharma, A.; Phang, J.M. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012, 72, 3677–3686.
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Hu, C.A.; Phang, J.M. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: The role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 2006, 25, 5640–5647.
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754.
- Stein, L.R.; Imai, S.-i. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428.
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258.
- Zhang, G.-F.; Jensen, M.V.; Gray, S.M.; El, K.; Wang, Y.; Lu, D.; Becker, T.C.; Campbell, J.E.; Newgard, C.B. Reductive TCA cycle metabolism fuels glutamine- and glucose-stimulated insulin secretion. Cell Metab. 2021, 33, 804–817.e5.
- Wang, Y.; Agarwal, E.; Bertolini, I.; Ghosh, J.C.; Seo, J.H.; Altieri, D.C. IDH2 reprograms mitochondrial dynamics in cancer through a HIF-1α-regulated pseudohypoxic state. FASEB J. 2019, 33, 13398–13411.
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065.
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658.


