The dynamics of oxidative stress navigates the cancer cell throughout its progression in different stages- where higher reactive oxygen species (ROS) favor malignant transformation, then decrease during tumor formation to the optimum level required for maintaining cellular redox homeostasis. It again increases during hypoxia and selects for aggressive metastatic cells. These cells are capable of withstanding the highly stressful steps of metastasis and the oxidative environment of the blood circulation. The redox status of the metastatic cells dictates successful colonization in distant organs [
1,
2,
3]. Technical challenges in their detection and modulation in vivo have greatly limited our understanding on their role [
4]. The dynamics of ROS during cancer progression are reciprocated in the level of TIGAR (TP53-induced glycolysis and apoptosis regulator), which is the P53-induced modulator for cellular adaptation to oxidative stress-increasing NADPH production and heightening the antioxidant defense. In pancreatic ductal adenocarcinoma development, during the early stages, high TIGAR levels are needed to mitigate the ROS associated with transformation. Then, as the tumor progresses, decreasing levels of TIGAR appear to increase the malignancy of cancer cells consistent with the selection for invasive cells with a higher ROS scavenging capacity. At a later stage, the TIGAR levels go up again to buffer the oxidative stress experienced by metastatic cells [
5]. Cancer cells need to develop higher ROS scavenging capabilities to maintain their required level for initiation, adaptation and progression and mitigate toxic accumulation [
6,
7,
8].
The alteration of oxidative stress is associated with the reprogramming of cellular metabolism during different stages of cancer progression to maintain redox homeostasis and support the metabolic demand imposed in each stage [
9]. Mitochondrial redox metabolism resides at the epicenter of this process and orchestrates different aspects of cellular metabolism for adaptation and, in association with generating ROS signaling actively, drives cancer cells’ progression through tumor development, metastasis and colonization in distant organs [
10,
11]. The role of mitochondria in cancer development is primarily focused on mutations in genes in TCA cycle enzymes, namely succinate dehydrogenase (SDH), fumarate hydratase (FH) and isocitrate dehydrogenase 2 (IDH2), leading to the accumulation of succinate, fumarate and 2-hydroxyglycerate (2HG). These metabolites mediate epigenetic changes leading to the development of some subtypes of cancer. When the mitochondrion is viewed in the context of its role in ROS signaling and reprogramming cellular metabolism, its impact is much more complex and substantial in different stages across all types of cancer [
12,
13,
14].
2. Mitochondrial Redox Metabolism
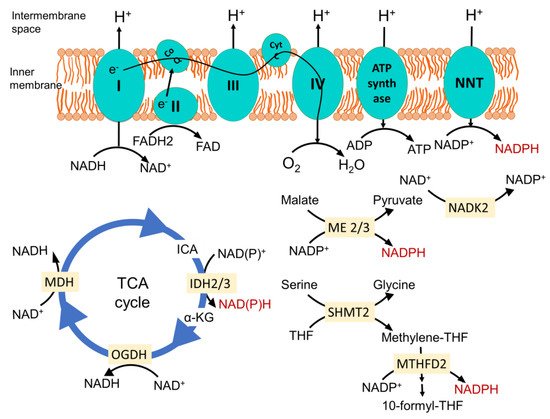
The mitochondrial electron transport chain (ETC)-mediated inner membrane electrochemical gradient drives oxidative phosphorylation for energy production. It is also the primary component for generating mitochondrial reactive oxygen species (ROS) and maintaining the redox balance (
Figure 1). In a situation where the sequential movement of electrons through the ETC is delayed or halted or ATP synthase is inhibited, creating increased membrane hyperpolarization, it causes the electrons at complexes I, II and III to interact with O
2 to form a superoxide [
2,
15]. Due to the structural organization, complex III is the primary site that can produce superoxide in the intermembrane space. The Fenton reaction produces a highly reactive hydroxyl radical via the reaction between Fe(II) and hydrogen per oxide (H
2O
2). This may interact with lipid species and generate lipid peroxyl radicals. Several enzymes in the mitochondria that participate in oxidation-reduction reactions, namely the 2-oxoglutarate dehydrogenase (OGDH), branched-chain 2-oxoacid dehydrogenase (BCKDH) and pyruvate dehydrogenase (PDH) complexes, may also contribute to ROS generation [
16].
Figure 1. Mitochondrial major NADPH-producing pathways. Nicotinamide nucleotide transhydrogenase (NNT) located in the inner membrane produces NAPDH from NADP+. It is energized by the inner membrane electrochemical gradient, produced by proton translocation via the electron transport chain (ETC) during the sequential flow of electrons from reducing equivalent NADH and FADH2 to O2 to form H2O. This enzyme plays a pivotal role in dedicating the mitochondrial NADH pool towards maintenance of the optimum NADPH level. Isocitrate dehydrogenase 2 (IDH2) in the TCA cycle catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate, and malic enzymes 2 and 3 (ME2 and ME3) catalyze the oxidative decarboxylation of malate to pyruvate while generating NADPH from NADP+ in the process. In the one carbon metabolism once serine is catabolized by serine hydroxymethyl transferase 2 (SHMT2), it generates methylene-THF. The NAD kinase (NADK2) phosphorylates NAD+ to NADP+, feeding other NADPH-generating reactions. Methylene tetrahydrofolate dehydrogenase 2 oxidizes it to methenyl tetrahydrofolate and, finally, to 10-formyl-THF, which is coupled to the reduction of NADP+ to NADPH. CoQ, coenzyme Q; Cyt C, cytochrome C; ICA, isocitrate; α-KG, α-ketoglutarate; OGDH, oxoglutarate dehydrogenase; MDH, malate dehydrogenase; THF, tetrahydrofolate; I, II, III and IV represents complexes I, II, III and IV in the mitochondrial electron transport chain.
ROS can also be produced in a regulated manner for signaling via NADPH oxidases (NOX proteins), which generates a superoxide from O
2 while oxidizing NADPH. Among seven catalytic isoforms, NOX1, NOX2, NOX4 and NOX5 are associated with cancer development, having different subcellular locations. NOX-derived ROS signaling induces proliferation upon growth factor stimulation and promotes angiogenesis, cell migration and invasion [
17]. NOX4 localized in the inner mitochondrial membrane can produce ROS, which can directly oxidize mitochondrial complex subunits, as well as mediate mitochondrial DNA damage [
18]. It also acts as an ATP sensor and mediates metabolic reprogramming via regulating pyruvate kinase-M2 (PKM2) stability [
19].
Mitochondrial superoxide is dismutated to H
2O
2 by manganese superoxide dismutase (MnSOD/SOD2) in the matrix and by Cu/Zn superoxide dismutase (Cu/ZnSOD/SOD1) in the intermembrane space and cytosol. H
2O
2 acts as diffusible signaling molecule and is neutralized for water by enzymes, including peroxiredoxin-thioredoxin and glutathione peroxidases (GPX) [
20]. Recent studies have drawn attention towards the pathological significance of mitochondrial peroxiredoxins, as their expression is associated with the initiation and progression of multiple types of cancers [
21]. These antioxidant systems depend on NADPH as the final electron donor, and hence, the redox balance of mitochondria relies on their NADP
+/NADPH ratio.
NADPH is primarily generated in mitochondria via mitochondrial inner membrane enzyme nicotinamide nucleotide transhydrogenase (NNT), which catalyzes the transfer of a hydride from NADH to NADP
+, producing NADPH and NAD
+, coupled to the translocation of one proton across the membrane (
Figure 1) [
22]. Since this enzyme is energized by a mitochondrial inner membrane electrochemical gradient and uses NADH as a reducing equivalent, it ensures the mitochondrial metabolism is dedicated to maintaining the optimum NADPH level in mitochondria required for antioxidant defense, as well as the biosynthesis of macromolecules. In a recent study, where they employed a genetically encoded tool to perturb the NAD
+/NADH and NADP
+/NADPH ratios in the mitochondria and cytosol, they found that the redox states of mitochondrial NADPH and NADH pools are connected, but they are asymmetrical. The oxidation of NADP
+/NADPH in the mitochondria leads to the oxidation of NAD
+/NADH, but the reverse is not true. This, along with the observation that perturbing the mitochondrial NADP
+/NADPH ratio increases the TCA cycle activity for the generation of NADH, indicates that the mitochondrial metabolism tends towards maintaining the optimum NADP
+/NAPDH ratio [
23]. Noteworthy to mention is that a similar connection has not been found for the cytosolic NADPH and NADH pools. Analyzing compartmentalized NADPH production revealed that NADH supports NADPH production in the mitochondria but not in the cytosol [
24]. Since NNT plays a direct role in consuming NADH to generate NADPH and NAD
+, it is the central player in integrating mitochondrial energy metabolism to maintain the redox balance. For this, NNT is considered a sensor of mitochondrial biology [
25,
26].
Other enzymes that play an important role in generating mitochondrial NADPH are mitochondrial TCA cycle enzyme isocitrate dehydrogenase 2 (IDH2), catalyzing the oxidative decarboxylation of isocitrate to α-ketoglutarate, and mitochondrial malic enzymes 2 and 3 (ME2 and ME3), catalyzing the oxidative decarboxylation of malate to pyruvate (
Figure 1) [
27,
28]. Serine catabolism via the mitochondrial one-carbon metabolism pathway is also important to generate NADPH (
Figure 1) [
29]. Mitochondrial NAD kinase (NADK2) phosphorylates NAD
+ to NADP
+, which is required for NADPH production. The loss of NADK2 is associated with reduced respiration and increased intracellular ROS, but its role in maintaining the redox balance and cancer is yet to be investigated [
30,
31]. One important concept that has emerged is that the maintenance of mitochondrial redox metabolism is not a separate event; rather, it is intricately tied with energy metabolism and biosynthetic processes and navigates metabolic reprogramming, bringing change to the mitochondrial biology and overall cell fate.
Sirtuin proteins, specifically SIRT3 residing in mitochondria, act as a key regulator of mitochondrial redox metabolism. It functions as a NAD
+-dependent protein deacetylase, which gets activated in high NAD
+/NADH conditions. Thus, it acts as a sensor of the mitochondrial metabolic state and activates pathways for maintaining the optimum ATP and NADPH levels [
32,
33,
34]. It reduces ROS generation from ETC and enhances the detoxification through the activation of antioxidant enzymes, like SOD2, resulting in maintenance of the redox balance in mitochondria [
35,
36,
37].
A critical role of mitochondrial redox metabolism is to support proline biosynthesis as it consumes NAD(P)H [
31]. NAD
+ generation during proline biosynthesis uncouples the TCA cycle from respiration and could help minimize the generation of ROS from ETC [
38]. The proline shuttle mediated by proline oxidase (POX) is important for transferring redox equivalents between the mitochondria and cytosol and generating ROS for signaling [
39,
40].
How far the cytosolic pathways support maintaining the redox balance in mitochondria is an area of active research. A compartmentalized NAD
+/NADH ratio is maintained in the mitochondria (NAD
+/NADH ratio 7 to 8) and cytosol (NAD
+/NADH ratio 60–700) mainly by malate–aspartate shuttle, malate–citrate shuttle and glycerophosphate shuttle [
41,
42]. Cytosolic reductive carboxylation comes into play for supporting mitochondrial NADPH generation for mitigating mitochondrial ROS. Here, IDH1 consumes NADPH from cytosol-generating citrate, which goes inside the mitochondria for oxidation, where IDH2 generates NADPH [
43]. This cycle can also work in reverse and follow the path of mitochondrial reductive carboxylation to support cytosolic NADPH production [
44]. In mitochondrial oxidative stress, the directionality of the cycle is maintained towards the generation of NADPH in the mitochondria through regulation of the IDH2 enzyme [
45].
In addition, mitochondrial plasticity comes into action when cells experience stress. Fusion helps to alleviate stress by complementing the content of two damaged mitochondria and, thus, maximize the oxidative capacity. Fission is needed to generate new mitochondria but, during stress, helps to remove damaged mitochondria as a measure of the quality control [
46]. Mitochondrial fission is also responsible for the generation of ROS in a hyperglycemic condition, and the induction of fusion can reduce ROS accumulation [
47].