 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Linda Borellini | + 2189 word(s) | 2189 | 2021-11-10 10:22:39 | | | |
| 2 | Jessie Wu | Meta information modification | 2189 | 2021-11-22 02:57:15 | | |
Video Upload Options
The progressive reduction of the dopaminergic neurons of the substantia nigra is the
fundamental process underlying Parkinson’s disease (PD), while the mechanism of susceptibility
of this specific neuronal population is largely unclear. Disturbances in mitochondrial function have
been recognized as one of the main pathways in sporadic PD since the finding of respiratory chain
impairment in animal models of PD. Studies on genetic forms of PD have provided new insight on
the role of mitochondrial bioenergetics, homeostasis, and autophagy. PINK1 (PTEN-induced
putative kinase 1) gene mutations, although rare, are the second most common cause of
recessively inherited early-onset PD, after Parkin gene mutations. Our knowledge of PINK1 and
Parkin function has increased dramatically in the last years, with the discovery that a process
called mitophagy, which plays a key role in the maintenance of mitochondrial health, is mediated
by the PINK1/Parkin pathway. In vitro and in vivo models have been developed, supporting the
role of PINK1 in synaptic transmission, particularly affecting dopaminergic neurons. It is of
paramount importance to further define the role of PINK1 in mitophagy and mitochondrial
homeostasis in PD pathogenesis in order to delineate novel therapeutic targets.
1. Background
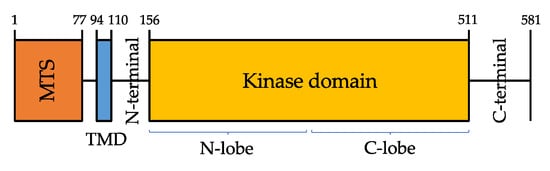
2. PINK1 Protein Functions
3. Clinical Features and Genotype-Phenotype Correlation
References
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13.
- Guzman, R.E.; Schwarz, Y.N.; Rettig, J.; Bruns, D. SNARE Force Synchronizes Synaptic Vesicle Fusion and Controls the Kinetics of Quantal Synaptic Transmission. J. Neurosci. 2010, 30, 10272–10281.
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology Of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87.
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803.
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273.
- Yamano, K.; Matsuda, N.; Tanaka, K. The ubiquitin signal and autophagy: An orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016, 17, 300–316.
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91.
- Winklhofer, K.F. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell Biol. 2014, 24, 332–341.
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429.
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101.
- Daou, S.; Sicheri, F. Vivid views of the PINK1 protein. Nat. Cell Biol. 2017, 552, 38–39.
- Kumar, A.; Tamjar, J.; Waddell, A.D.; Woodroof, H.I.; Raimi, O.G.; Shaw, A.M.; Peggie, M.; Muqit, M.M.; Van Aalten, D.M. Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. eLife 2017, 6, e29985.
- Schubert, A.F.; Gladkova, C.; Pardon, E.; Wagstaff, J.L.; Freund, S.M.V.; Steyaert, J.; Maslen, S.L.; Komander, D. Structure of PINK1 in complex with its substrate ubiquitin. Nat. Cell Biol. 2017, 552, 51–56.
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517.
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520.
- Langston, J.W.; Ballard, P.A. Parkinson’s Disease in a Chemist Working with 1-Methyl-4-Phenyl-L,2,5,6-Tetrahydropyridine. N. Engl. J. Med. 1983, 309, 310.
- Schapira, A.; Cooper, J.; Dexter, D.; Jenner, P.; Clark, J.; Marsden, C. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 333, 1269.
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385.
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.; Whitworth, A.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2010, 20, 867–879.
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769.
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333.
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080.
- Chen, Y.; Dorn, G.W., II. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475.
- Tanaka, K. The PINK1–Parkin axis: An Overview. Neurosci. Res. 2020, 159, 9–15.
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153.
- Kazlauskaite, A.; Martinez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1-dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954.
- Sauvé, V.; Lilov, A.; Seirafi, M.; Vranas, M.; Rasool, S.; Kozlov, G.; Sprules, T.; Wang, J.; Trempe, J.; Gehring, K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015, 34, 2492–2505.
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nat. Cell Biol. 2014, 510, 162–166.
- Wang, L.; Cho, Y.-L.; Tang, Y.; Wang, J.; Park, J.-E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1–Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802.
- Thomas, R.E.; Andrews, L.A.; Burman, J.L.; Lin, W.-Y.; Pallanck, L.J. PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix. PLoS Genet. 2014, 10, e1004279.
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The Mitochondrial Fusion-Promoting Factor Mitofusin Is a Substrate of the PINK1/Parkin Pathway. PLoS ONE 2010, 5, e10054.
- Gegg, M.E.; Schapira, A.H. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011, 7, 243–245.
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111.
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207.
- Voigt, A.; Berlemann, L.A.; Winklhofer, K.F. The mitochondrial kinase PINK1: Functions beyond mitophagy. J. Neurochem. 2016, 139, 232–239.
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A Vesicular Transport Pathway Shuttles Cargo from Mitochondria to Lysosomes. Curr. Biol. 2012, 22, 135–141.
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H. Vps35 Mediates Vesicle Transport between the Mitochondria and Peroxisomes. Curr. Biol. 2010, 20, 1310–1315.
- Vilariño-Güell, C.; Wider, C.; Ross, O.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 162–167.
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A Mutation in VPS35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 168–175.
- Charman, M.; Kennedy, B.E.; Osborne, N.; Karten, B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res. 2010, 51, 1023–1034.
- Elbaz-Alon, Y.; Rosenfeld-Gur, E.; Shinder, V.; Futerman, A.; Geiger, T.; Schuldiner, M. A Dynamic Interface between Vacuoles and Mitochondria in Yeast. Dev. Cell 2014, 30, 95–102.
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386.
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276.
- Todkar, K.; Chikhi, L.; Germain, M. Mitochondrial interaction with the endosomal compartment in endocytosis and mitochondrial transfer. Mitochondrion 2019, 49, 284–288.
- Petit, A.; Kawarai, T.; Paitel, E.; Sanjo, N.; Maj, M.; Scheid, M.; Chen, F.; Gu, Y.; Hasegawa, H.; Salehi-Rad, S.; et al. Wild-Type PINK1 Prevents Basal and Induced Neuronal Apoptosis, a Protective Effect Abrogated by Parkinson Disease-Related Mutations. J. Biol. Chem. 2005, 280, 34025–34032.
- Klinkenberg, M.; Thurow, N.; Gispert, S.; Ricciardi, F.; Eich, F.; Prehn, J.; Auburger, G.; Kögel, D. Enhanced vulnerability of PARK6 patient skin fibroblasts to apoptosis induced by proteasomal stress. Neuroscience 2010, 166, 422–434.
- Haque, M.E.; Mount, M.P.; Safarpour, F.; Abdel-Messih, E.; Callaghan, S.; Mazerolle, C.; Kitada, T.; Slack, R.; Wallace, V.; Shen, J.; et al. Inactivation of Pink1 Gene in Vivo Sensitizes Dopamine-Producing Neurons to 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and Can Be Rescued by Autosomal Recessive Parkinson Disease Genes, Parkin or DJ-1. J. Biol. Chem. 2012, 287, 23162–23170.
- Geisler, S.; Holmström, K.; Skujat, D.; Fiesel, F.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131.
- Bonifati, V.; Rohe, C.F.; Breedveld, G.J.; Fabrizio, E.; De Mari, M.; Tassorelli, C.; Tavella, A.; Marconi, R.; Nicholl, D.J.; Chien, H.F.; et al. Early-onset parkinsonism associated with PINK1 mutations: Frequency, genotypes, and phenotypes. Neurology 2005, 65, 87–95.
- Ricciardi, L.; Petrucci, S.; Guidubaldi, A.; Ialongo, T.; Serra, L.; Ferraris, A.; Spanò, B.; Bozzali, M.; Valente, E.M.; Bentivoglio, A.R. Phenotypic variability of PINK1 expression: 12 Years’ clinical follow-up of two Italian families. Mov. Disord. 2014, 29, 1561–1566.
- Piredda, R.; Desmarais, P.; Masellis, M.; Gasca-Salas, C. Cognitive and psychiatric symptoms in genetically determined Parkinson’s disease: A systematic review. Eur. J. Neurol. 2019, 27, 229–234.
- Nishioka, K.; Kefi, M.; Jasinska-Myga, B.; Wider, C.; Vilarino-Guell, C.; Ross, O.; Heckman, M.G.; Middleton, L.T.; Ishihara-Paul, L.; Gibson, R.; et al. A comparative study of LRRK2, PINK1 and genetically undefined familial Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2009, 81, 391–395.
- Choi, J.M.; Woo, M.S.; Ma, H.-I.; Kang, S.Y.; Sung, Y.-H.; Yong, S.W.; Chung, S.J.; Kim, J.-S.; Shin, H.-W.; Lyoo, C.H.; et al. Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 2008, 9, 263–269.
- Weng, Y.-H.; Chou, Y.-H.W.; Wu, W.-S.; Lin, K.-J.; Chang, H.-C.; Yen, T.-C.; Chen, R.-S.; Wey, S.-P.; Lu, C.-S. PINK1 mutation in Taiwanese early-onset parkinsonism. J. Neurol. 2007, 254, 1347–1355.
- Brooks, J.; Ding, J.; Sánchez, J.S.; Paisan-Ruiz, C.; Singleton, A.B.; Scholz, S. Parkin and PINK1 mutations in early-onset Parkinson’s disease: Comprehensive screening in publicly available cases and control. J. Med. Genet. 2009, 46, 375–381.
- Hedrich, K.; Hagenah, J.; Djarmati, A.; Hiller, A.; Lohnau, T.; Lasek, K.; Grünewald, A.; Hilker, R.; Steinlechner, S.; Boston, H.; et al. Clinical Spectrum of Homozygous and Heterozygous PINK1 Mutations in a Large German Family with Parkinson Disease. Arch. Neurol. 2006, 63, 833–838.
- Puschmann, A.; Fiesel, F.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117.
- Krohn, L.; Grenn, F.P.; Makarious, M.B.; Kim, J.J.; Bandres-Ciga, S.; Roosen, D.A.; Gan-Or, Z.; Nalls, M.A.; Singleton, A.B.; Blauwendraat, C. Comprehensive assessment of PINK1 variants in Parkinson’s disease. Neurobiol. Aging 2020, 91, 168.e1–168.e5.
- Schneider, S.A.; Klein, C. PINK1 Type of Young-Onset Parkinson Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.

