Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vera de Wit-Verheggen | + 2690 word(s) | 2690 | 2021-11-15 04:39:14 | | | |
| 2 | Peter Tang | Meta information modification | 2690 | 2021-11-18 02:38:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
De Wit-Verheggen, V. Changes in Cardiac Metabolism in Prediabetes. Encyclopedia. Available online: https://encyclopedia.pub/entry/16096 (accessed on 25 July 2026).
De Wit-Verheggen V. Changes in Cardiac Metabolism in Prediabetes. Encyclopedia. Available at: https://encyclopedia.pub/entry/16096. Accessed July 25, 2026.
De Wit-Verheggen, Vera. "Changes in Cardiac Metabolism in Prediabetes" Encyclopedia, https://encyclopedia.pub/entry/16096 (accessed July 25, 2026).
De Wit-Verheggen, V. (2021, November 17). Changes in Cardiac Metabolism in Prediabetes. In Encyclopedia. https://encyclopedia.pub/entry/16096
De Wit-Verheggen, Vera. "Changes in Cardiac Metabolism in Prediabetes." Encyclopedia. Web. 17 November, 2021.
Copy Citation
In type 2 diabetes mellitus (T2DM), there is an increased prevalence of cardiovascular disease (CVD), even when corrected for atherosclerosis and other CVD risk factors. Diastolic dysfunction is one of the early changes in cardiac function that precedes the onset of cardiac failure, and it occurs already in the prediabetic state.
cardiac metabolism
prediabetes
mitochondrial function
cardiac function
1. Introduction
Prediabetes, defined as impaired fasting glucose (fasting plasma glucose between 6.1 and 6.9 mmol/L) or impaired glucose tolerance (2-h plasma glucose between 7.8 and 11.0 mmol/L) [1], places individuals at high risk of developing type 2 diabetes mellitus (T2DM) and its cardiovascular disease (CVD)-related complications [2][3]. The increased risk of CVD is proportional to the fasting blood glucose in prediabetes [2][4][5] and is mainly caused by atherosclerosis, induced by the many risk factors that are characteristic for prediabetic patients, for instance, dyslipidemia and hypertension [6][7][8][9][10][11][12]. Atherosclerosis leading to ischemic heart disease has been extensively discussed in previous literature [13]. However, even when corrected for atherosclerosis, cholesterol values, bodyweight, blood pressure, and age, patients with prediabetes remain at increased risk for the development of heart failure, mainly through the development of diastolic dysfunction (in T2DM known as diabetic cardiomyopathy (DCM)) [14][15]. This phenomenon is also part of the spectrum better known as heart failure with a preserved ejection fraction (HFpEF) [16].
Interestingly, diastolic dysfunction has been shown to be present not only in T2DM but also in prediabetes [17]. Evidence associates higher glucose levels with lower cardiac function parameters in prediabetes [17], indicating that changes in cardiac function arise early in the development of T2DM. Changes in cardiac metabolism in response to hyperglycemia are considered to be an important pathway through which T2DM causes DCM [18][19], and this may already be at play in prediabetes. Recognition of these metabolic changes may help to better understand the underlying etiology of diastolic dysfunction in prediabetes, which provides a window of opportunity for the prevention of DCM in the early development of T2DM.
2. Cardiac Fat
When energy intake exceeds expenditure, it eventually results in body fat accumulation, as can be seen in obesity [20]. The large surplus of nutrients also leads to the development of fat deposits in organs other than adipose tissue, such as skeletal muscle, the liver, and the heart. Such ectopic lipid accumulation has been related to insulin resistance in many tissues. Thus, cardiac fat accumulation may play an important role in the development of diastolic changes in the prediabetic heart. Cardiac lipid content can be studied by both an in vivo and an ex vivo approach.
In vivo studies use magnetic resonance spectroscopy (MRS) for the relative quantification of metabolites involved in lipid metabolism. Proton MRS (1H-MRS) generates a spectrum wherein multiple lipid signals (CH2 and CH3), a creatine (Cr) signal, and a water (H2O) signal can be distinguished. CH2/H2O is generally used as a parameter that reflects myocardial triglyceride content, and this mainly represents neutral lipid storage as triglycerides in the myocardium. With this in vivo technique, it was shown that the myocardial triglyceride content is increased in overweight and obese individuals [21] and possibly even more in prediabetes and T2DM [22], in comparison to lean individuals. In addition, myocardial triglyceride content was weakly associated with insulin sensitivity, as determined by the homeostasis model assessment index [22].
Ex vivo studies found increased intramyocardial lipid deposition in patients with metabolic syndrome (average HOMA score 4.2 ± standard deviation 0.5, which often is considered as prediabetes) [23], obesity or T2DM [24], compared to lean patients. In addition, Anderson et al. showed that in nondiabetic and diabetic individuals, the cardiac fat content correlated positively with the HbA1c [25]. These ex vivo studies suggest that cardiac fat already in prediabetic individuals is increased compared to healthy lean individuals [23][24][25], which is in line with the above-mentioned in vivo results of McGavock et al. [22].
Interestingly, several links between cardiac fat accumulation and cardiac function have been described. Van der Meer et al. showed that in lean individuals with a normal glucose metabolism (NGM), an increased myocardial triglyceride content (following a very low-calorie diet) measured by 1H-MRS was correlated with a decrease in diastolic function [26]. In addition, in individuals with metabolic syndrome (and a high HOMA score), a correlation was found between the amount of cardiac fat accumulation and the progression of cardiac dysfunction (measured by myocardial performance index and ejection fraction) [23]. The increased myocardial triglyceride content in overweight and obese individuals was accompanied by elevated LV mass and suppressed septal wall thickening as measured by cardiac imaging, compared to lean individuals [21]. This suggests that possibly in prediabetes, an increased cardiac fat storage may influence cardiac function negatively.
3. Adipose Tissue Surrounding the Heart
The fat deposits around the heart (epicardial adipose tissue and pericardial adipose tissue) also typically increase with overweight/obesity and are reported to be more pronounced in diabetic patients. These depots have not been measured specifically in prediabetic populations, and although specific data of these separate depots in prediabetes are lacking, it might be expected that the epicardial adipose tissue (EAT) is elevated in prediabetes since cardiac fat is increased in prediabetes and the thickness of the EAT is strongly correlated with the cardiac fat in healthy males [27]. However, quantitative studies are needed to confirm this concept in prediabetic individuals.
From obese individuals, it is known that when EAT expands, the balance between the storage and release of fatty acids shifts toward a more active secretion [28]. Furthermore, the expanded EAT transforms its secretory profile toward more pro-inflammatory cytokines and chemokines, negatively affecting neighboring cells [29][30][31]. This results in a chronic inflammatory response that is shown to be present in enlarged EAT tissue [32][33]. Moreover, this local secretion of inflammatory mediators can also inhibit the activity of insulin. Indeed, EAT is positively associated with insulin resistance and metabolic syndrome [34][35].
4. Enhanced Cardiac Lipid Metabolism
Insulin usually inhibits lipolysis and thereby reduces the release of plasma non-esterified fatty acids (NEFAs). However, in individuals with reduced insulin sensitivity, as is the case in prediabetes, the postprandial effect of insulin is impaired, leaving the circulating free fatty acids (FFA) elevated [36]. In addition to the increased FFA levels in the circulation, PET studies show that both the FFA uptake and the FFA oxidation in the prediabetic myocardium are increased. Using 18F-fluoro-6-thia-heptadecanoic acid (FTHA) as a fatty acid tracer and [11C]acetate to determine cardiac perfusion and oxidative metabolic index, Labbé et al. showed that in prediabetic individuals (defined as impaired glucose tolerance), an increased NEFA uptake in the heart and an increased myocardial oxidative metabolism for the first 6 h postprandially compared to the individuals with a normal glucose metabolism (NGM) [37]. This was in contrast to the uptake of fatty acids in liver and skeletal muscle since these remained similar in prediabetes compared to NGM in the postprandial state [37]. These findings concerning increased FFA availability in the plasma and myocardial FFA metabolism are confirmed by Brassard et al. in normoglycemic first-degree relatives of T2DM individuals (who are therefore at highly increased risk to develop T2DM) in comparison to matched individuals having no increased risk for T2DM. Using the stable isotopic tracers ([1,1,2,3,3-2H5]-glycerol and [U-13C]-palmitate or [1,2-13C]-acetate), they showed that these individuals at high risk for T2DM during enhanced intravascular TG lipolysis at high insulin levels have both an increased plasma appearance of NEFAs and increased myocardial oxidation of the NEFAs [36].
These findings in the insulin-stimulated condition from Brassard and Labbé point out that already in prediabetes, changes in cardiac fatty acid handling occur, with increased uptake and oxidation of fatty acids in the heart in comparison to NGM. Moreover, Labbé et al. revealed that these changes in lipid metabolism may be maladaptive regarding cardiac function. The increased uptake and oxidation of NEFA in the prediabetic individuals was associated with a reduced left ventricular ejection fraction (LVEF), reduced left ventricular stroke volume, and tended to display impaired diastolic function [37]. This is in line with the findings from Mather et al. in T2DM individuals, who showed that the augmented myocardial fatty acid oxidation under fasted and insulin-treated conditions (measured by16-[18F]fluoro-4-thiapalmitate (FTP) and 11C-acetate) was accompanied by reduced cardiac work efficiency [38]. This may not be surprising since increased fatty acid oxidation at the expense of carbohydrate oxidation increases oxygen demand, resulting in reduced myocardial efficiency [38]. In addition, in prediabetic individuals with a known increased risk for atherosclerosis, this makes the heart more prone to ischemia. The enhanced fatty acid metabolism in prediabetes has, therefore, implications for contractile performance and ischemia tolerance [38].
It may be beneficial to counterbalance this altered substrate metabolism in order to prevent DCM in T2DM. The metabolic changes that occur early on in the prediabetic heart seem to be reversible, as shown by several studies in prediabetic individuals. Six months after bariatric surgery, individuals with prediabetes showed an improvement in whole-body insulin sensitivity, which correlated positively with the decrease in myocardial fasting free fatty acid uptake, but also myocardial function. Although cardiac fat was not reduced, the myocardial structure was improved [39]. Similar results in prediabetes were observed by Labbé et al., where modest weight loss following a 1-year lifestyle intervention led to changes in substrate metabolism and improved cardiac function [40]. However, a short-term diet of 7 days in prediabetes did not achieve these improvements in cardiac function [41] and thus suggesting that structural changes regarding cardiac metabolism and function take longer to develop.
5. Decreased Cardiac Glucose Metabolism
Together with alterations in cardiac fatty acid metabolism, reciprocal changes in cardiac glucose metabolism may be expected in prediabetes [42]. Here, PET studies using the glucose analog [18F]-fluorodeoxyglucose (18F-FDG) can provide insight into the myocardial uptake of glucose. Kim et al. studied a mixed population of NGM, prediabetes, and T2DM and revealed that the visceral fat area and fasting FFA are independent determinants of myocardial glucose uptake in the fasted condition [43]. However, both Kim et al. and Hu et al. showed that prediabetes was not associated with decreased myocardial glucose uptake in a fasted condition, whereas T2DM was [43][44], which is in line with animal studies [45]. However, findings might be different in a fed or insulin-stimulated state.
In contrast to the fasted individuals in the study of Kim et al., Nielsen et al. studied the myocardial glucose uptake 1 h after oral glucose intake in NGM, prediabetes, and newly diagnosed T2DM individuals, all characterized by chronic heart failure and reduced LVEF. Even though the myocardial blood flow and myocardial flow reserve were similar, individuals with prediabetes and newly diagnosed T2DM had-despite of elevated levels of glucose and insulin-a decreased myocardial glucose uptake compared to NGM [46]. However, since the authors did not separate analyses for individuals with prediabetes and T2DM, it is unknown whether there were differences between these groups.
To assess the insulin-stimulated myocardial glucose uptake in a more controlled setting than right after glucose ingestion, one should measure myocardial glucose uptake during a hyperinsulinemic euglycemic clamp [47]. Eriksson et al. showed a similar cardiac glucose metabolic rate during such clamp in control, prediabetes, and T2DM individuals matched for age, sex, and BMI [48]. Others showed lower myocardial glucose uptake in T2DM compared to (BMI-matched overweight) NGM individuals during a hyperinsulinemic euglycemic clamp [47][49]. These conflicting in vivo findings of myocardial glucose uptake in T2DM are also found in ex vivo studies. Full-thickness myocardial biopsies from the left ventricle of T2DM individuals showed an increase in cardiac insulin receptor substrate 1 (IRS1)–PI 3-kinases (PI3K) activity compared to their overweight controls with NGM [50]. This means that the insulin signaling cascade, even in this state of insulin resistance, is intact. However, differences between groups can be blunted due to the fact that all individuals in the ex vivo studies were characterized by left ventricular dysfunction.
Overall, results on myocardial glucose uptake in prediabetes are conflicting, both in in vivo and ex vivo studies. Some found no differences in healthy prediabetic individuals compared to NGM or T2DM individuals in a fasted state [43] and during a clamp [48], whereas others did find reduced myocardial glucose uptake 1 h after oral glucose loading [46] in prediabetic patients with chronic heart failure. In addition, previous literature is ambiguous whether myocardial glucose uptake is associated with whole-body insulin sensitivity [49] or not [48] measured during a hyperinsulinemic euglycemic clamp. Data are dispersed, and the question remains whether prediabetes is characterized by reduced myocardial insulin sensitivity.
6. Mitochondrial Function
Mitochondria are responsible for oxidative metabolism and are key to the normal function of the cardiomyocytes. It is, therefore, not surprising that mitochondrial dysfunction is suspected of playing a pivotal role in the development of DCM [51][52]. Unfortunately, human data in prediabetes in this area are lacking. Though studies in male Long-Evans rats that were high-fat-fed and had a streptozotocin treatment as a model for prediabetes have shown a mild diastolic dysfunction and cardiac hypertrophy associated with early changes in mitophagy [53]. This supports the suggestion that mitochondrial dysfunction underlies the development of DCM in prediabetes.
However, information from both in vivo and ex vivo studies performed in obesity and T2DM is available and may explain possible changes in mitochondrial function in prediabetes. From ex vivo studies using high-resolution respirometry as a reflection of mitochondrial function, it is known that lower mitochondrial respiration is associated with T2DM [25][54].
In vivo, concentrations of high-energy phosphates have been suggested to be closely related to cardiac mitochondrial oxidation. This can be measured by 31P-MRS. With this technique, PCr and ATP can be recognized at a specific resonance frequency. As a relative quantification, the ratio of PCr over ATP is used as a measure for myocardial energy status. Mitochondria produce ATP during oxidative phosphorylation, and ATP can, in turn, be used to convert creatine (Cr) into PCr. In the sarcolemma, the phosphate group of PCr is exchanged with adenosine diphosphate (ADP) to form ATP in case of increased energy demand. In this way, PCr acts to buffer ATP. This PCr shuttle system is also shown in Figure 1. In the normal myocardium, ATP synthesis can be maintained at the rate of ATP demand, and PCr levels are sustained. However, in cardiac disease with a decreased mitochondrial function, ATP demand may outweigh the mitochondrial capacity for ATP production, and hence, PCr concentrations will fall [55]. Hence, the PCr/ATP ratio has been suggested to be a marker of mitochondrial function; however, one should be aware that creatine supply, pH, and oxygen supply may independently influence PCr concentrations in the cardiomyocyte [56].
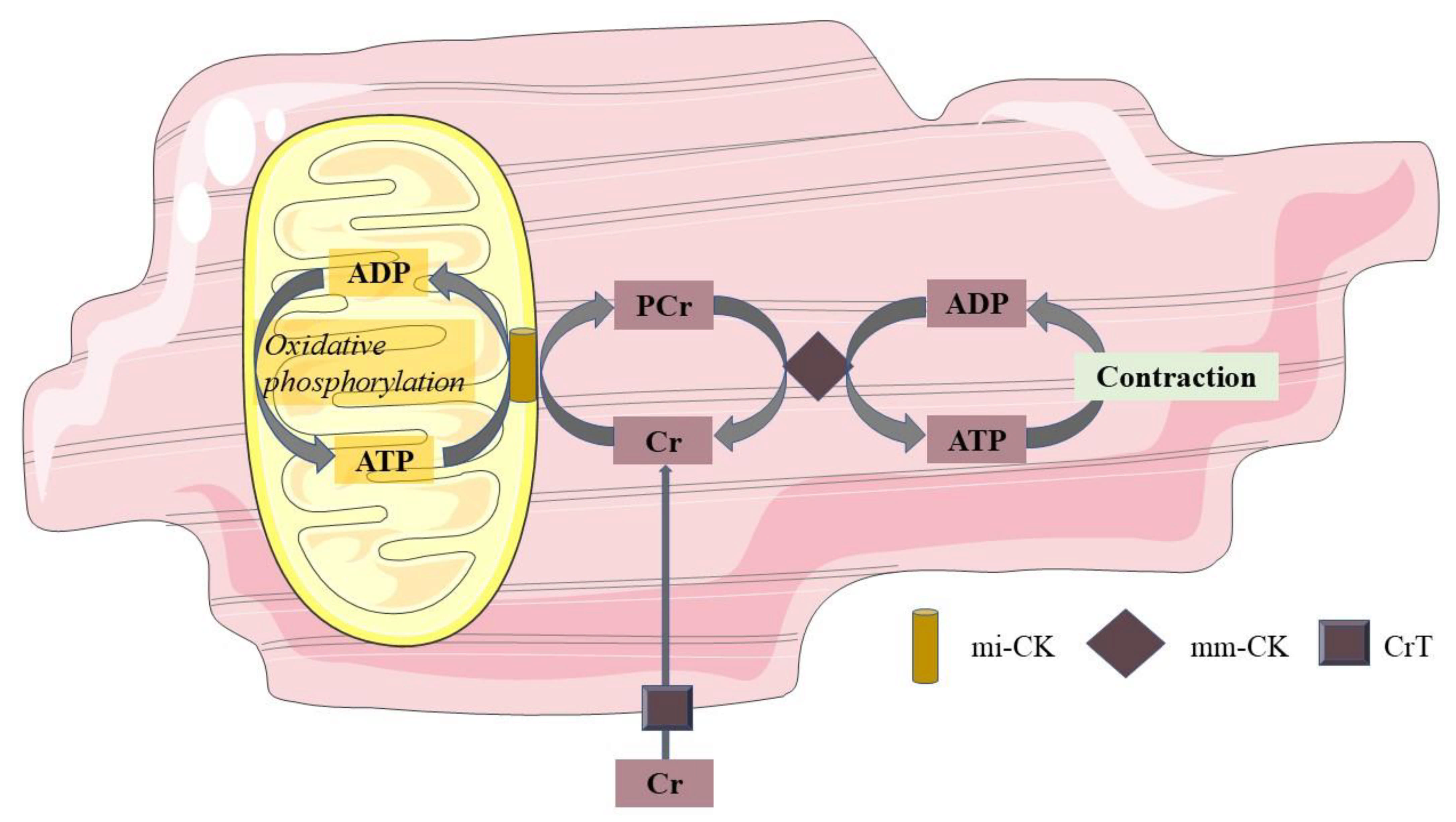
Figure 1. Phosphocreatine shuttle system. Mitochondria produce ATP during oxidative phosphorylation, and this ATP converts Cr at the mitochondrial membrane into PCr through mi-CK. PCr, in turn, shuttles from the mitochondrial membrane to the sarcolemma, where through the mm-CK, the phosphorous group of PCr is exchanged with ADP to form ATP in cases of increased energy demand. In this way, PCr acts to buffer ATP. ADP adenosine diphosphate; ATP adenosine triphosphate; Cr free creatinine; PCr phosphocreatinine; mi-CK mitochondrial creatine kinase; mm-CK myofibrillar creatine kinase; CrT creatinine transporter.
Secondly, gene regulatory pathways may affect mitochondrial function by influencing the interplay between the supply and oxidation of the various substrates (Figure 2). Not only in individuals with T2DM but also in first-degree relatives of individuals with T2DM, it has been shown that the expression of peroxisome proliferator-activated receptor (PPAR) coactivator PGC-1α is decreased in skeletal muscle [57]. PGC-1 is usually increased upon cellular ATP demand [58], leading to transcription of NRF-1, PPARα, and PPARγ, and may thereby have indirect effects on mitochondrial metabolism. Since NRF-1 regulates the expression of many mitochondrial genes, including the OXPHOS genes, a decreased expression of PGC-1α will result in a lower mitochondrial content of the OXPHOS complexes. These data found in skeletal muscle may be translated to heart, though data in prediabetes are lacking. Yet, in T2DM, Montaigne et al. found a downregulation of NRF-1 and mitochondrial function in the heart [54].
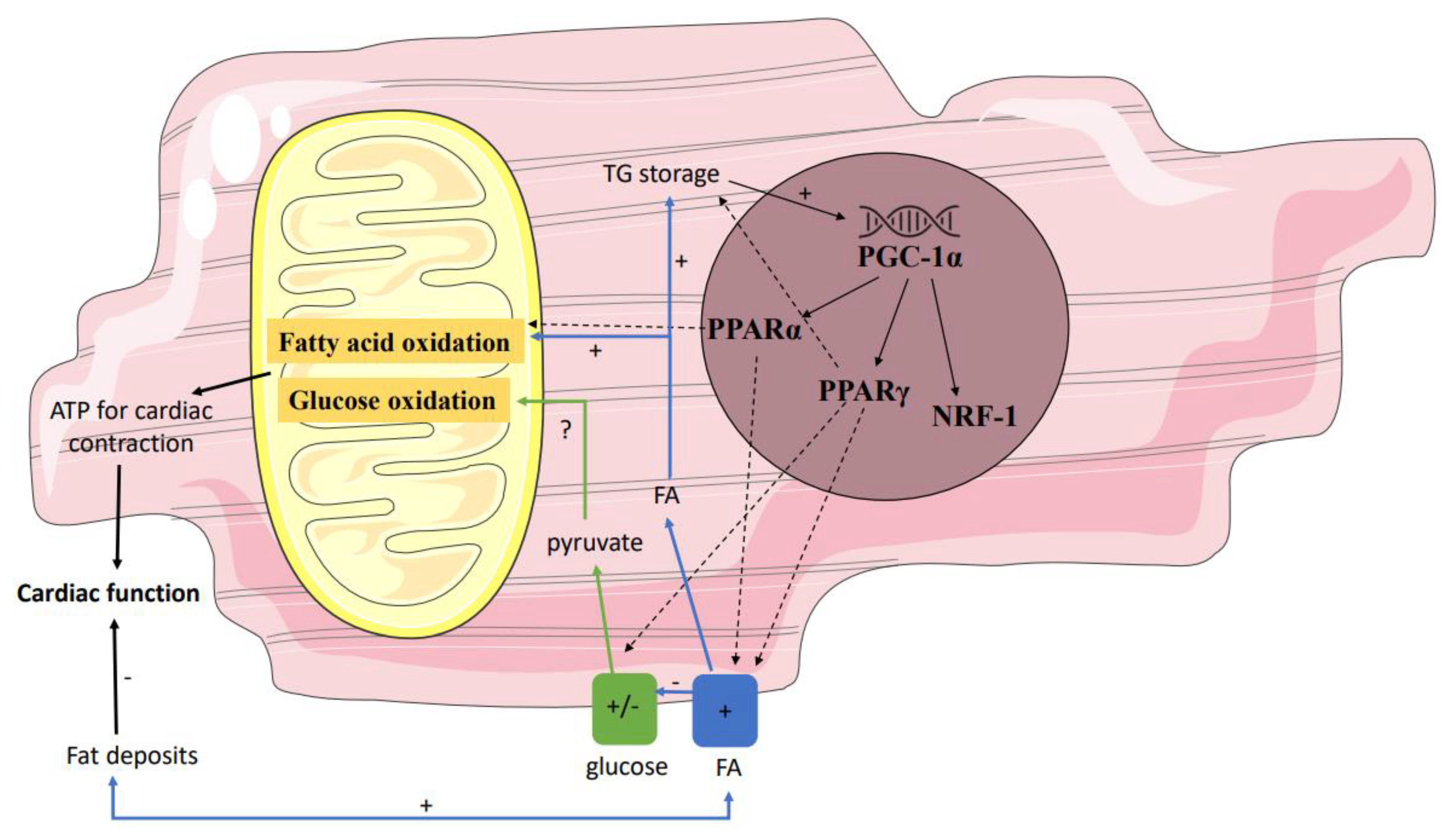
Figure 2. Gene regulatory pathways affecting mitochondrial function. Several genes influence the interplay between the supply and oxidation of the various substrates. Down- and/or upregulation of these genes in prediabetes may affect mitochondrial function. Partly because of this, in prediabetes fatty acid oxidation may be stimulated, resulting in a net reduction in ATP and thus reduced myocardial efficiency. PPAR peroxisome proliferator-activated receptor; ATP adenosine triphosphate.
References
- World Health Organization; International Diabetes Federation. Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycemia: Report of a WHO/IDF Consultation: WHO. 2006. Available online: https://www.who.int/diabetes/publications/Definition%20and%20diagnosis%20of%20diabetes_new.pdf (accessed on 12 October 2021).
- Punthakee, Z.; Goldenberg, R.; Katz, P. Definition, Classification and Diagnosis of Diabetes, Prediabetes and Metabolic Syndrome. Can. J. Diabetes 2018, 42 (Suppl. S1), S8–S11.
- Cai, X.; Zhang, Y.; Li, M.; Wu, J.H.; Mai, L.; Li, J.; Yang, Y.; Hu, Y.; Huang, Y. Association between prediabetes and risk of all cause mortality and cardiovascular disease: Updated meta-analysis. BMJ 2020, 370, m2297.
- Haffner, S.M.; Mykkanen, L.; Festa, A.; Burke, J.P.; Stern, M.P. Insulin-resistant prediabetic subjects have more atherogenic risk factors than insulin-sensitive prediabetic subjects: Implications for preventing coronary heart disease during the prediabetic state. Circulation 2000, 101, 975–980.
- Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222.
- Grundy, S.M.; Benjamin, I.J.; Burke, G.L.; Chait, A.; Eckel, R.H.; Howard, B.V.; Mitch, W.; Smith, S.C., Jr.; Sowers, J.R. Diabetes and cardiovascular disease: A statement for healthcare professionals from the American Heart Association. Circulation 1999, 100, 1134–1146.
- Hammoud, T.; Tanguay, J.F.; Bourassa, M.G. Management of coronary artery disease: Therapeutic options in patients with diabetes. J. Am. Coll. Cardiol. 2000, 36, 355–365.
- Taegtmeyer, H.; McNulty, P.; Young, M.E. Adaptation and maladaptation of the heart in diabetes: Part I: General concepts. Circulation 2002, 105, 1727–1733.
- Eckel, R.H.; York, D.A.; Rossner, S.; Hubbard, V.; Caterson, I.; St Jeor, S.T.; Hayman, L.L.; Mullis, R.M.; Blair, S.N. Prevention Conference VII: Obesity, a worldwide epidemic related to heart disease and stroke: Executive summary. Circulation 2004, 110, 2968–2975.
- Calle, E.E.; Thun, M.J.; Petrelli, J.M.; Rodriguez, C.; Heath, C.W., Jr. Body-mass index and mortality in a prospective cohort of U.S. adults. N. Engl. J. Med. 1999, 341, 1097–10105.
- Wolk, R.; Berger, P.; Lennon, R.J.; Brilakis, E.S.; Davison, D.E.; Somers, V.K. Association between plasma adiponectin levels and unstable coronary syndromes. Eur. Heart J. 2007, 28, 292–298.
- Tirosh, A.; Shai, I.; Afek, A.; Dubnov-Raz, G.; Ayalon, N.; Gordon, B.; Derazne, E.; Tzur, D.; Shamis, A.; Vinker, S.; et al. Adolescent BMI trajectory and risk of diabetes versus coronary disease. N. Engl. J. Med. 2011, 364, 1315–1325.
- Lloyd-Jones, D.; Adams, R.J.; Brown, T.M.; Carnethon, M.; Dai, S.; De Simone, G.; Ferguson, T.B.; Ford, E.; Furie, K.; Gillespie, C.; et al. Executive summary: Heart disease and stroke statistics—2010 update: A report from the American Heart Association. Circulation 2010, 121, 948–954.
- Van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18.
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34.
- LeWinter, M.M.; Meyer, M. Mechanisms of diastolic dysfunction in heart failure with a preserved ejection fraction: If it’s not one thing it’s another. Circ. Heart Fail. 2013, 6, 1112–1115.
- Markus, M.R.P.; Rospleszcz, S.; Ittermann, T.; Baumeister, S.E.; Schipf, S.; Siewert-Markus, U.; Lorbeer, R.; Storz, C.; Ptushkina, V.; Peters, A.; et al. Glucose and insulin levels are associated with arterial stiffness and concentric remodeling of the heart. Cardiovasc. Diabetol. 2019, 18, 145.
- Bell, D.S. Diabetic cardiomyopathy. Diabetes Care 2003, 26, 2949–2951.
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671.
- Roberts, S.B. Abnormalities of energy expenditure and the development of obesity. Obes. Res. 1995, 3 (Suppl. S2), 155s–163s.
- Szczepaniak, L.S.; Dobbins, R.L.; Metzger, G.J.; Sartoni-D’Ambrosia, G.; Arbique, D.; Vongpatanasin, W.; Unger, R.; Victor, R.G. Myocardial triglycerides and systolic function in humans: In vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn. Reson. Med. 2003, 49, 417–423.
- McGavock, J.M.; Lingvay, I.; Zib, I.; Tillery, T.; Salas, N.; Unger, R.; Levine, B.D.; Raskin, P.; Victor, R.G.; Szczepaniak, L.S. Cardiac steatosis in diabetes mellitus: A 1H-magnetic resonance spectroscopy study. Circulation 2007, 116, 1170–1175.
- Marfella, R.; Di Filippo, C.; Portoghese, M.; Barbieri, M.; Ferraraccio, F.; Siniscalchi, M.; Cacciapuoti, F.; Rossi, F.; D’Amico, M.; Paolisso, G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J. Lipid Res. 2009, 50, 2314–2323.
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700.
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898.
- Van der Meer, R.W.; Hammer, S.; Smit, J.W.; Frolich, M.; Bax, J.J.; Diamant, M.; de Roos, A.; Romijn, J.A.; Lamb, H.J. Short-term caloric restriction induces accumulation of myocardial triglycerides and decreases left ventricular diastolic function in healthy subjects. Diabetes 2007, 56, 2849–2853.
- Malavazos, A.E.; Di Leo, G.; Secchi, F.; Lupo, E.N.; Dogliotti, G.; Coman, C.; Morricone, L.; Corsi, M.M.; Sardanelli, F.; Iacobellis, G. Relation of echocardiographic epicardial fat thickness and myocardial fat. Am. J. Cardiol. 2010, 105, 1831–1835.
- Bakkum, M.J.; Danad, I.; Romijn, M.A.; Stuijfzand, W.J.; Leonora, R.M.; Tulevski, I.I.; Somsen, G.A.; Lammertsma, A.A.; van Kuijk, C.; van Rossum, A.C.; et al. The impact of obesity on the relationship between epicardial adipose tissue, left ventricular mass and coronary microvascular function. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1562–1573.
- Cherian, S.; Lopaschuk, G.D.; Carvalho, E. Cellular cross-talk between epicardial adipose tissue and myocardium in relation to the pathogenesis of cardiovascular disease. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E937–E949.
- Gaborit, B.; Abdesselam, I.; Dutour, A. Epicardial fat: More than just an “epi” phenomenon? Horm. Metab. Res. 2013, 45, 991–1001.
- Iozzo, P. Myocardial, perivascular, and epicardial fat. Diabetes Care 2011, 34 (Suppl. S2), S371–S379.
- Henrichot, E.; Juge-Aubry, C.E.; Pernin, A.; Pache, J.C.; Velebit, V.; Dayer, J.M.; Meda, P.; Chizzolini, C.; Meier, C.A. Production of chemokines by perivascular adipose tissue: A role in the pathogenesis of atherosclerosis? Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2594–2599.
- Shibasaki, I.; Nishikimi, T.; Mochizuki, Y.; Yamada, Y.; Yoshitatsu, M.; Inoue, Y.; Kuwata, T.; Ogawa, H.; Tsuchiya, G.; Ishimitsu, T.; et al. Greater expression of inflammatory cytokines, adrenomedullin, and natriuretic peptide receptor-C in epicardial adipose tissue in coronary artery disease. Regul. Pept. 2010, 165, 210–217.
- Iacobellis, G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat. Rev. Endocrinol. 2015, 11, 363–371.
- Iacobellis, G.; Willens, H.J. Echocardiographic epicardial fat: A review of research and clinical applications. J. Am. Soc. Echocardiogr. 2009, 22, 1311–1319.
- Brassard, P.; Frisch, F.; Lavoie, F.; Cyr, D.; Bourbonnais, A.; Cunnane, S.C.; Soinio, M.; Dadson, P.; Virtanen, K.A.; Gronroos, T.; et al. Impaired plasma nonesterified fatty acid tolerance is an early defect in the natural history of type 2 diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 837–844.
- Labbe, S.M.; Grenier-Larouche, T.; Noll, C.; Phoenix, S.; Guerin, B.; Turcotte, E.E.; Carpentier, A.C. Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes 2012, 61, 2701–2710.
- Mather, K.J.; Hutchins, G.D.; Perry, K.; Territo, W.; Chisholm, R.; Acton, A.; Glick-Wilson, B.; Considine, R.V.; Moberly, S.; DeGrado, T.R. Assessment of myocardial metabolic flexibility and work efficiency in human type 2 diabetes using 16-fluoro-4-thiapalmitate, a novel PET fatty acid tracer. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E452–E460.
- Hannukainen, J.C.; Lautamäki, R.; Pärkkä, J.; Strandberg, M.; Saunavaara, V.; Hurme, S.; Soinio, M.; Dadson, P.; Virtanen, K.A.; Grönroos, T.; et al. Reversibility of Myocardial Metabolism and Remodeling in Morbidly Obese Patients Six Months after Bariatric Surgery. Diabetes Obes. Metab. 2018, 20, 963–973.
- Labbe, S.M.; Noll, C.; Grenier-Larouche, T.; Kunach, M.; Bouffard, L.; Phoenix, S.; Guerin, B.; Baillargeon, J.P.; Langlois, M.F.; Turcotte, E.E.; et al. Improved cardiac function and dietary fatty acid metabolism after modest weight loss in subjects with impaired glucose tolerance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1388–E1396.
- Noll, C.; Kunach, M.; Frisch, F.; Bouffard, L.; Dubreuil, S.; Jean-Denis, F.; Phoenix, S.; Cunnane, S.C.; Guerin, B.; Turcotte, E.E.; et al. Seven-Day Caloric and Saturated Fat Restriction Increases Myocardial Dietary Fatty Acid Partitioning in Impaired Glucose-Tolerant Subjects. Diabetes 2015, 64, 3690–3699.
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin resistance: Metabolic mechanisms and consequences in the heart. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2068–2076.
- Kim, G.; Jo, K.; Kim, K.J.; Lee, Y.H.; Han, E.; Yoon, H.J.; Wang, H.J.; Kang, E.S.; Yun, M. Visceral adiposity is associated with altered myocardial glucose uptake measured by (18)FDG-PET in 346 subjects with normal glucose tolerance, prediabetes, and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 148.
- Hu, L.; Qiu, C.; Wang, X.; Xu, M.; Shao, X.; Wang, Y. The association between diabetes mellitus and reduction in myocardial glucose uptake: A population-based 18F-FDG PET/CT study. BMC Cardiovasc. Disord. 2018, 18, 203.
- Van den Brom, C.E.; Huisman, M.C.; Vlasblom, R.; Boontje, N.M.; Duijst, S.; Lubberink, M.; Molthoff, C.F.; Lammertsma, A.A.; van der Velden, J.; Boer, C.; et al. Altered myocardial substrate metabolism is associated with myocardial dysfunction in early diabetic cardiomyopathy in rats: Studies using positron emission tomography. Cardiovasc. Diabetol. 2009, 8, 39.
- Nielsen, R.; Jorsal, A.; Iversen, P.; Tolbod, L.; Bouchelouche, K.; Sorensen, J.; Harms, H.J.; Flyvbjerg, A.; Botker, H.E.; Wiggers, H. Heart failure patients with prediabetes and newly diagnosed diabetes display abnormalities in myocardial metabolism. J. Nucl. Cardiol. 2018, 25, 169–176.
- Ohtake, T.; Yokoyama, I.; Watanabe, T.; Momose, T.; Serezawa, T.; Nishikawa, J.; Sasaki, Y. Myocardial glucose metabolism in noninsulin-dependent diabetes mellitus patients evaluated by FDG-PET. J. Nucl. Med. 1995, 36, 456–463.
- Eriksson, J.W.; Visvanathar, R.; Kullberg, J.; Strand, R.; Skrtic, S.; Ekström, S.; Lubberink, M.; Lundqvist, M.H.; Katsogiannos, P.; Pereira, M.J.; et al. Tissue-specific glucose partitioning and fat content in prediabetes and type 2 diabetes: Whole-body PET/MRI during hyperinsulinemia. Eur. J. Endocrinol. 2021, 184, 879–889.
- Yokoyama, I.; Yonekura, K.; Ohtake, T.; Kawamura, H.; Matsumoto, A.; Inoue, Y.; Aoyagi, T.; Sugiura, S.; Omata, M.; Ohtomo, K.; et al. Role of insulin resistance in heart and skeletal muscle F-18 fluorodeoxyglucose uptake in patients with non-insulin-dependent diabetes mellitus. J. Nucl. Cardiol. 2000, 7, 242–248.
- Cook, S.A.; Varela-Carver, A.; Mongillo, M.; Kleinert, C.; Khan, M.T.; Leccisotti, L.; Strickland, N.; Matsui, T.; Das, S.; Rosenzweig, A.; et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur. Heart J. 2010, 31, 100–111.
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414.
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153.
- Koncsos, G.; Varga, Z.V.; Baranyai, T.; Boengler, K.; Rohrbach, S.; Li, L.; Borra, R.; Härkönen, R.; Iozzo, P.; Stewart, M.; et al. Diastolic dysfunction in prediabetic male rats: Role of mitochondrial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H927–H943.
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.M.; Mouton, S.; Sebti, Y.; et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 2014, 130, 554–564.
- Van de Weijer, T.; Paiman, E.H.M.; Lamb, H.J. Mini-Review on Cardiac Metabolic Imaging: Current imaging modalities and future perspectives. J. Appl. Physiol. 2018, 124, 168–181.
- McMahon, S.; Jenkins, D. Factors affecting the rate of phosphocreatine resynthesis following intense exercise. Sports Med. 2002, 32, 761–784.
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471.
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
18 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No