Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Margaret Brimble | + 2787 word(s) | 2787 | 2021-10-14 08:14:02 | | | |
| 2 | Camila Xu | + 27 word(s) | 2814 | 2021-11-16 08:02:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Brimble, M. FDA Approved Antibody-Drug Conjugates. Encyclopedia. Available online: https://encyclopedia.pub/entry/16031 (accessed on 05 August 2026).
Brimble M. FDA Approved Antibody-Drug Conjugates. Encyclopedia. Available at: https://encyclopedia.pub/entry/16031. Accessed August 05, 2026.
Brimble, Margaret. "FDA Approved Antibody-Drug Conjugates" Encyclopedia, https://encyclopedia.pub/entry/16031 (accessed August 05, 2026).
Brimble, M. (2021, November 16). FDA Approved Antibody-Drug Conjugates. In Encyclopedia. https://encyclopedia.pub/entry/16031
Brimble, Margaret. "FDA Approved Antibody-Drug Conjugates." Encyclopedia. Web. 16 November, 2021.
Copy Citation
Antibody-drug conjugate (ADC) are now amongst the fastest growing drug classes in oncology, as they combine the best features of mAbs and small molecule drugs, creating a single moiety that is highly specific and cytotoxic.
antibody-drug conjugates
ADCs
targeted therapy
cancer
FDA approved
1. Introduction
Paul Ehrlich’s vision of a rationally targeted strategy to eliminate disease, whether it be microbes or malignant cells, has driven research over the past century, particularly creating a targeted cancer therapy revolution [1]. In 1913, it was theorized that a so-called ‘magic bullet’ drug could cause selective destruction by employing a toxin and a targeting agent. Over 80 years following Ehrlich’s fundamental realization, and supported by the successful development of chemotherapy in the 1940s [2] and monoclonal antibodies (mAbs) in the 1970s [3], in 1983 the first successful antibody-drug conjugate (ADC) human clinical trial began using an anti-carcinoembryonic antibody tethered to vindesine [4]. The safety of administration and the ability of the conjugate to localize after radiolabeling was investigated in eight patients with advanced metastatic carcinoma. While the feasibility of this approach was demonstrated, several hurdles were identified, the most significant being aggregation [4].
ADCs are now amongst the fastest growing drug classes in oncology, as they combine the best features of mAbs and small molecule drugs, creating a single moiety that is highly specific and cytotoxic. These therapeutic entities are considered the “homing missiles” of cancer therapy, and are composed of three key elements: a monoclonal antibody that selectively binds to an antigen on the tumor cell surface, a cytotoxic drug payload, and a cleavable or non-cleavable linker, see Figure 1 [5][6][7]. Each of these components can vary widely between ADCs, leading to immense diversity in the overall structure, and subsequently, the ADC’s pharmacological and clinical properties. ADCs are designed to deliver the toxic payload selectively to cells expressing the target antigen. Therefore, target antigens that are preferentially expressed in tumors versus non-malignant cells can be exploited to harness a greater therapeutic window and reduce the chance of off-target effects associated with systemic administration of traditional chemotherapeutics. The advent of ADCs has thus sparked a revival of chemotherapeutic payloads, which cannot be administered systemically due to their extreme potency and ensuing toxicity profiles.
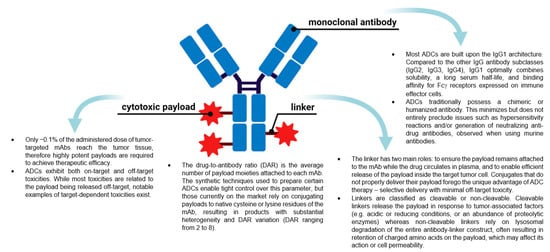
Figure 1. The general structure of an antibody-drug conjugate (ADC) and key points about the different components. (Created with BioRender.com, accessed 27 September 2021).
Many ADCs have demonstrated impressive activity against treatment-refractory cancers, resulting in their approval for both hematologic malignancies and solid tumor indications. At the time of writing, 11 different ADCs have been approved by the US Food and Drug Administration (FDA) for clinical use, see Figure 2A and Table 1. Of these, seven have also obtained approval by the European Medicines Agency (EMA) (Appendix A). The recent surge in ADC approvals, of which Polivy® (polatuzumab vedotin-piiq), Padcev® (enfortumab vedotin-ejfv), Enhertu® (fam-trastuzumab deruxtecan-nxki), Trodelvy® (sacituzumab govitecan-hziy), Blenrep® (belantamab mafodotin-blmf), Zynlonta® (loncastuximab tesirine-lpyl), and Tivdak® (tisotumab vedotin-tftv) have all gained FDA approval since 2019, belies the turbulent past these biologics have experienced, both in academic and regulatory settings.
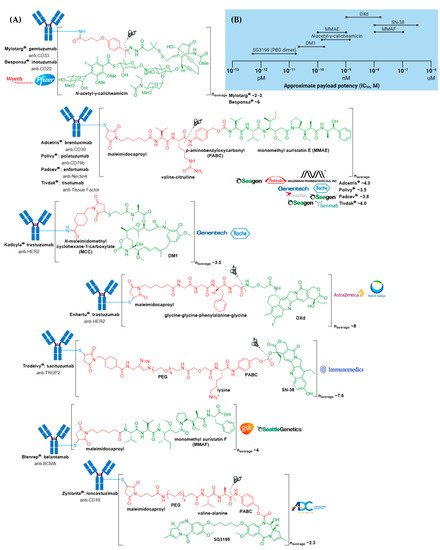
Figure 2. (A) Structures of FDA approved antibody-drug conjugates (ADCs). The antibody is shown in blue, and chemical structures for linker and payload are in red and green, respectively. Scissors indicate the cleavage site (if applicable). Pharmaceutical makers and drug-to-antibody ratio for each ADC is indicated. (B) Comparison of approximate payload potency ranges (Created with BioRender.com, accessed September 2021).
Table 1. FDA approved ADCs currently on the market.
| ADC | Target | mAb | Linker | Payload/Payload Class | Payload Action | DAR | Disease Indication (Year of Approval) |
|---|---|---|---|---|---|---|---|
| Mylotarg® (gemtuzumab ozogamicin) | CD33 | IgG4 | acid cleavable | ozogamicin/calicheamicin | DNA cleavage | 2–3 | CD33+ R/R AML (2000) a |
| Adcetris® (brentuximab vedotin) | CD30 | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 4 | R/R sALCL or cHL (2011)R/R pcALCL or CD30+ MF (2017); cHL, sALCL or CD30+ PTCL (2018) b |
| Kadcyla® (ado-trastuzumab emtansine) | HER2 | IgG1 | non-cleavable | DM1/maytansinoid | microtubule inhibitor | 3.5 | HER2+ metastatic breast cancer previously treated with trastuzumab & a taxane (2013); HER2+ early breast cancer after neoadjuvant taxane & trastuzumab-based treatment (2019) |
| Besponsa® (inotuzumab ozogamicin) | CD22 | IgG4 | acid cleavable | ozogamicin/calicheamicin | DNA cleavage | 6 | R/R B-ALL (2017) |
| Polivy® (polatuzumab vedotin-piiq) | CD79b | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 3.5 | R/R DLBCL (2019) c,d |
| Padcev® (enfortumab vedotin-ejfv) | Nectin4 | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 3.8 | Locally advanced or metastatic urothelial cancer after a PD-1 or PD-L1 inhibitor and a Pt-containing chemotherapy (2019) or are ineligible for cisplatin-containing chemotherapy and previously received 1 or more lines of therapy (2021) d |
| Enhertu® (fam-trastuzumab deruxtecan-nxki) | HER2 | IgG1 | enzyme cleavable | DXd/camptothecin | TOP1 inhibitor | 8 | Unresectable or metastatic HER2+ breast cancer after 2 or more anti-HER2 regimens (2019) d; locally advanced or metastatic HER2+ gastric or gastroesophageal junction adenocarcinoma after a trastuzumab-based regimen (2021) |
| Trodelvy® (sacituzumab govitecan-hziy) | TROP2 | IgG1 | acid cleavable | SN-38/camptothecin | TOP1 inhibitor | 7.6 | Locally advanced or metastatic TNBC after at least two prior therapies (2020); locally advanced or metastatic urothelial cancer after a Pt-containing chemotherapy and a PD-1 or PD-L1 inhibitor (2021) d |
| Blenrep® (belantamab mafodotin-blmf) | BCMA | IgG1 | non-cleavable | MMAF/auristatin | microtubule inhibitor | 4 | R/R multiple myeloma after at least 4 prior therapies including an anti-CD38 mAb, a proteasome inhibitor, and an immunomodulatory agent (2020) d |
| Zynlonta® (loncastuximab tesirine-lpyl) | CD19 | IgG1 | enzyme cleavable | SG3199/PBD dimer | DNA cleavage | 2.3 | R/R large B-cell lymphoma after 2 or more lines of systemic therapy, including DLBCL not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma (2021) d |
| Tivdak® (tisotumab vedotin-tftv) | Tissue Factor | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 4 | Recurrent or metastatic cervical cancer with disease progression on or after chemotherapy (2021) d |
ADC, antibody-drug conjugate; AML, acute myeloid leukemia; B-ALL, B-cell acute lymphoblastic leukemia; BCMA, B-cell maturation antigen; cHL, classical Hodgkin lymphoma; DAR, drug-to-antibody ratio; DLBCL, diffuse large B-cell lymphoma; mAb, monoclonal antibody; MF, mycosis fungoides; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; pcALCL, primary cutaneous anaplastic large cell lymphoma; Pt, platinum; PTCL, peripheral T-cell lymphoma; PBD, pyrrolobenzodiazepine; R/R, relapsed and/or refractory; sALCL, systemic anaplastic large cell lymphoma; TOP1, topoisomerase I; TROP2, tumor-associated calcium signal transducer 2. aAs a single agent or in combination with daunorubicin and cytarabine. Mylotarg® was withdrawn from the market in 2010 and reapproved in 2017 for newly diagnosed R/R CD33-positive AML. b In combination with cyclophosphamide, doxorubicin, and prednisone for newly diagnosed sALCL or CD30+ PTCL and in combination with doxorubicin, vinblastine, and dacarbazine for newly diagnosed cHL. c In combination with bendamustine and rituximab. d Indication approved under accelerated approval.
While several publications have listed Lumoxiti® (moxetumomab pasudotox-tdfk) as an FDA approved ADC [8][9], we have excluded it from our discussions as we consider it an immunotoxin [10][11][12][13]. Besides Lumoxiti® [14][15], the immunotoxins Ontak® (denileukin diffittox) [16] and Elzonris® (tagraxofusp-erzs) [17], have also been granted FDA approval.
2. ADC Mechanism of Action
The general mechanism of action for an ADC is depicted in Figure 3. Following the introduction of the ADC into the plasma circulation (step 1), recognition of a specific antigen on the tumor cell surface leads to strong binding and formation of an antigen–ADC complex (step 2). The entire complex is internalized, predominantly through receptor-mediated endocytosis with formation of a clathrin-coated early endosome (step 3) [18]. Inside the early endosome, some ADCs bind neonatal Fc receptors (FcRns) and undergo transcytosis to the extracellular space (step 4a) [18][19]. Following endosomal maturation to a late endosome, characterized by an environment with low luminal pH [20], those ADCs retained in the endosome undergo drug release from cleavable linkers (step 4b). The late endosome fuses with a lysosome (step 5), inside which the ADC and/or its components are exposed to proteolytic enzymes (e.g., cathepsin B) and an increasingly acidic environment, promoting further payload release (step 6). The free drug then exerts its cellular destruction via a pathway specific to the mode of action of the payload. Most ADC payloads cause apoptosis by DNA damage or microtubule disruption (step 7). In addition, some payloads (those sufficiently hydrophobic to cross cell membranes and initially tethered to an antibody via a cleavable linker) exert a bystander effect. Free drug is exported from the target tumor cell, across the cell membrane to kill neighboring tumor cells, including those that may not express the relevant antigen on its cell surface or are less accessible directly from the circulatory system (step 8).
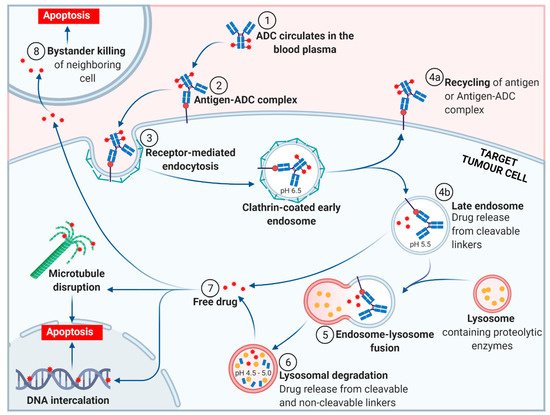
Figure 3. The general mechanism of action of an antibody-drug conjugate (ADC). (Adapted from “Antibody-Drug Conjugate Release”, by BioRender.com (accessed 27 September 2021). Retrieved from https://app.biorender-templates, accessed 27 September 2021).
3. FDA Approved ADCs
3.1. Mylotarg®
Mylotarg® (gemtuzumab ozogamicin) from Wyeth/Pfizer was the first ADC to reach the market. It is composed of a recombinant humanized anti-CD33 mAb (IgG4κ antibody hP67.6) covalently attached to a calicheamicin derived payload (N-acetyl-γ-calicheamicin 1,2-dimethyl hydrazine dichloride) via a pH-sensitive hydrazone linker, see Figure 4 [21][22].
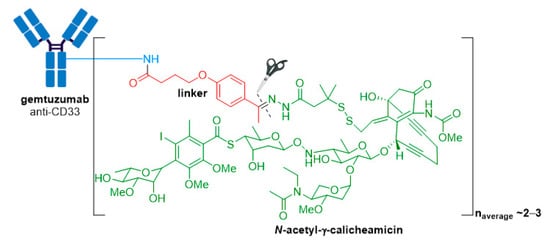
Figure 4. Structure for Mylotarg® (gemtuzumab ozogamicin). The antibody is shown in blue, and chemical structures for linker and payload are in red and green, respectively. The cleavage site is indicated by scissors.
Highlighting the rocky start for ADC therapeutics, Mylotarg® was granted accelerated approval for relapsed CD33+ acute myeloid leukemia (AML) in 2000, but was voluntarily withdrawn from the market in 2010 after post-approval studies failed to verify survival benefit and demonstrated a higher rate of fatal toxicity than chemotherapy alone [23][24]. Despite this, Mylotarg® was reapproved by the FDA in 2017 under an alternative dosing regimen (previously administered as one dose of 9 mg/m2) of three doses of 3 mg/m2, and a different patient population was introduced [25]. These changes reduced the maximum plasma concentration, thus improving the safety profile and response rate when administered as a single-agent [26][27] or combination regimen [28][29].
The antitumor activity of Mylotarg® results from the semi-synthetic payload, a calicheamicin derivative (N-acetyl-γ-calicheamicin 1,2-dimethyl hydrazine dichloride) produced by microbial fermentation followed by synthetic modification. The payload consists of four glycosidic units, a fully substituted iodobenzoate moiety, and an aglycon. The highly reactive hex-3-ene-1,5-diyne subunit can be readily triggered to aromatize via a Bergman cyclization reaction, generating a benzene-1,4-diradical [30]. This aromatization process affords a resulting diradical that can abstract two hydrogen atoms from the DNA backbone, leading to unrepairable double-strand (ds) DNA breaks followed by cell-cycle arrest and apoptotic cell death, see Figure 5 [30].
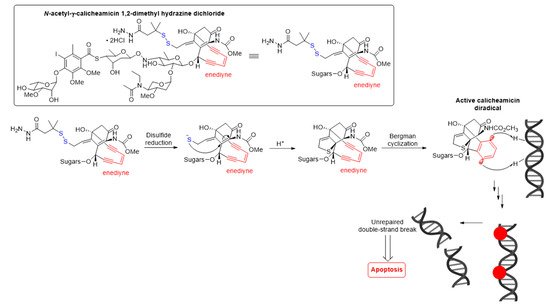
Figure 5. Mechanism for double-strand (ds) DNA cleavage by N-acetyl-γ-calicheamicin. The enediyne warhead is shown in red.
A crucial feature for successful construction of an ADC is the conjugation chemistry of the linker-payload with the mAb. In Mylotarg®, the bifunctional 4-(4-acetylphenoxy)butanoic acid moiety provides attachment to surface-exposed lysine residues of the mAb through an amide bond, and the linker forms an acyl hydrazone linkage with the payload. Mylotarg® is considered a first-generation ADC because it utilizes N-hydroxysuccinimide chemistry to conjugate calicheamicin to surface-exposed lysine residues on the antibody, yielding a heterogenous mixture with different drug-to-antibody ratios (DARs) [31]. The number of conjugated calicheamicin derivatives per mAb ranges from zero to six, with an average drug loading of two to three molecules of calicheamicin per antibody.
The acid-cleavable hydrazone linker is designed to be stable in the neutral pH conditions encountered during circulation, however, hydrolysis is readily achieved under the acidic environment of lysosomes (pH ~4.5–5.0) inside CD33+ target cells. The dimethyl disulfide moiety preserves the natural disulfide trigger mechanism of calicheamicin, while the added steric hindrance resulting from the methyl substituents protects the disulfide from reduction during circulation [31][32].
As for all humanized antibodies, complementarity determining region (CDR) grafting was used for humanization of the anti-CD33 murine antibody, hP67.6, employed in Mylotarg® [33][34]. The resulting antibody is a genetically engineered IgG4κ antibody containing sequences derived from the murine antibody, but with an increased similarity to antibody variants produced naturally in humans. While the IgG4 antibody isotype has the longest circulating half-life of all isotypes, it is least likely to participate in immune-mediated mechanisms, such as complement fixation and antibody-dependent cellular cytotoxicity (ADCC) [35]. Although antibody effector functions, such as ADCC, complement-dependent cytotoxicity, and antibody-dependent cellular phagocytosis (ADCP), have the potential to augment antitumor activities, engaging Fcγ receptors can also lead to increased off-target and dose-limiting toxicity [36][37][38]. Several next-generation ADCs have thus exploited antibody engineering to enhance or impair immune effector functions.
Demonstrating that failure is perhaps merely a step towards success, the pitfalls and limitations of this first-generation ADC provided several key lessons for future improvements in ADC research.
3.2. Adcetris®
Adcetris® (brentuximab vedotin) from Seagen (formerly Seattle Genetics), containing a CD30-specific mAb conjugated to monomethyl auristatin E (MMAE), received FDA approval in 2011, making it the second ADC to enter the oncology market, see Figure 6 [39][40][41][42]. It is approved for Hodgkin lymphoma (HL) [43][44] and systemic anaplastic large cell lymphoma (sALCL) [45] in the USA, Europe, and Japan [40][46].
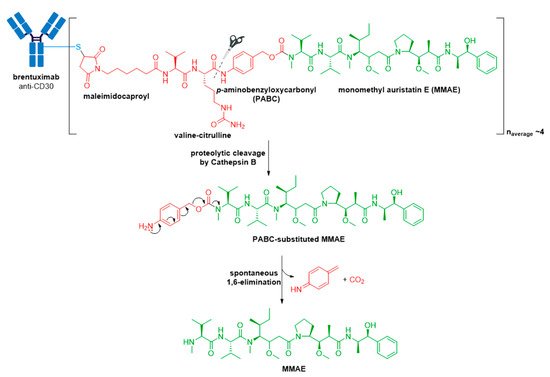
Figure 6. Structure of Adcetris® (brentuximab vedotin). The antibody is shown in blue, and chemical structures for linker and payload are in red and green, respectively. Spontaneous 1,6-elimination mechanism for the PABC-substituted MMAE, leading to release of MMAE, p-iminoquinone methide, and carbon dioxide.
The anticancer activity of Adcetris® results from the binding of MMAE to tubulin. This disrupts the microtubule network within the cell, subsequently inducing cell cycle arrest and apoptotic cell death [47]. In addition, likely owing to the IgG1 antibody isotype, in vitro data provide evidence for ADCP antitumor activity [48]. From first-generation ADCs, it was learnt that ~0.1% of the injected ADC dose reaches the target tumor site, thus necessitating an increase in potency of the cytotoxic agent and/or DAR for improved therapeutic activity [49][50]. Adcetris® addressed these two requirements by employing the more cytotoxic payload MMAE, a tubulin-targeting agent, belonging to the auristatin family of drug payloads (cytotoxicity in the low nanomolar to sub-nanomolar range against a variety of cancer types). See Figure 2B for a comparison of approximate cytotoxicity ranges (based on concentrations giving 50% maximum inhibition, IC50) for payloads employed in FDA approved ADCs. Furthermore, as compared to Mylotarg® with a DAR of two to three, Adcetris® has approximately four molecules of MMAE attached to each antibody molecule.
The pitfall of premature drug release resulting from the acid-cleavable hydrazone linker in Mylotarg® [51] was addressed in the second-generation ADC, Adcetris®, by using the protease-cleavable “mc-vc-PABC-MMAE” linker-drug combination [41][42][52][53][54]. This linker construct utilizes a thiol-reactive maleimidocaproyl (mc) spacer, a valine-citrulline (vc) dipeptide, and a self-immolative para-aminobenzyloxycarbonyl (PABC) spacer [54]. The mc spacer is incorporated for conjugation to cysteine residues of the mAb, and a PABC spacer allows linker attachment to the secondary amine of MMAE. Due to the steric bulk of the payload, the PABC spacer also facilitates enzyme access allowing the vc group to be recognized by cathepsin B [51][54][55]. Cathepsin B is a cysteine protease which presents almost exclusively in the lysosomal compartment in healthy mammals, and is overexpressed in multiple cancer types [56][57]. It is responsible for cleaving the citrulline-PABC amide bond. Following proteolytic cleavage, the resultant PABC-substituted MMAE forms an unstable intermediate which spontaneously undergoes a 1,6-elimination with loss of p-iminoquinone methide and carbon dioxide to release the free drug, see Figure 6.
Compared to Mylotarg®, which uses an IgG4 antibody, the IgG subclass employed in Adcetris® is IgG1. This is the most common subclass for ADCs, as while having similarly long serum half-lives to IgG4, they possess greater complement-fixation and FcγR-binding efficiencies [35].
Although Mylotarg® utilizes lysine residues on the mAb for linker-payload conjugation, Adcetris® employs cysteine-based conjugation. Due to the limited number of cysteine conjugation sites available (four interchain and twelve intrachain disulfides, see Figure 7, as opposed to 80–100 lysine amines for IgG1) and the distinct reactivity of thiols, this approach enables improved homogeneity of the ADC species and a more controlled drug loading [58]. Cysteine conjugation relies on partial or full reduction of the four interchain disulfides to produce an average number (e.g., two, four, six, or eight) of free nucleophilic thiols, while keeping the intrachain disulfide bonds intact. Interchain disulfides are generally not critical for structural stability and have higher solvent accessibility, making them an ideal target. They are typically reduced using reagents such as tris(2-carboxyethyl)phosphine (TCEP), dithiothreitol (DTT), or 2-mercaptoethylamine (2-MEA) prior to conjugation [58]. Once the free thiols are generated, they can be reacted with a linker-payload complex possessing a suitable electrophilic group, see Figure 7. Maleimide chemistry has been the mainstay for linkage to cysteines, with all auristatin-containing ADCs utilizing the maleimidocaproyl (mc) linkage to the antibody [55].
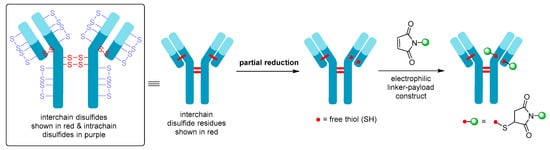
Figure 7. Schematic showing partial reduction of IgG1 antibody interchain disulfide bonds to generate two nucleophilic free thiol groups that can be reacted with an electrophilic linker-payload construct, such as maleimide (DAR = 2). Maleimide conjugation to cysteine is shown in this example.
Although an improvement over lysine conjugation, this method still produces a heterogenous mixture of ADC species, which can negatively impact on parameters including pharmacokinetics, tolerability, and efficacy [59]. Therefore, site-specific conjugation methodologies have been developed, of which THIOMABTM technology is the most well-known [60][61]. Genentech’s THIOMABTM antibody platform uses site-directed mutagenesis to incorporate cysteine residues into antibodies at positions on light and heavy chains that provide reactive thiol groups without perturbing immunoglobulin folding and assembly, or altering antigen binding [60][61]. Although homogenous ADCs have repeatedly demonstrated superior overall pharmacological profiles compared to their heterogenous counterparts, engineered antibodies for site-specific conjugation have not yet been employed in any of the FDA approved ADCs. We recommend the review by Walsh and co-workers for an in-depth understanding of chemical and enzymatic methods for site-specific antibody modification, resulting in the generation of homogenous ADCs [58].
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480.
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653.
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497.
- Ford, C.H.; Newman, E.C.; Johnson, J.R.; Woodhouse, C.S.; Reeder, A.T.; Rowland, G.F.; Simmonds, R.G. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 1983, 47, 35–42.
- Tolcher, A.W. The evolution of antibody-drug conjugates: A positive inflexion point. Am. Soc. Clin. Oncol. Educ. Book 2020, 127–134.
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244.
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29.
- Pazo, C.D.; Nawaz, K.; Webster, R.M. The oncology market for antibody–drug conjugates. Nat. Rev. Drug Discov. 2021, 20, 583–584.
- Kostova, V.; Désos, P.; Starck, J.-B.; Kotschy, A. The chemistry behind ADCs. Pharmaceuticals 2021, 14, 442.
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883.
- Mazor, R.; Pastan, I. Immunogenicity of immunotoxins containing pseudomonas exotoxin A: Causes, consequences, and mitigation. Front. Immunol. 2020, 11, 1261.
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565.
- Bruins, W.S.C.; Zweegman, S.; Mutis, T.; Van De Donk, N.W.C.J. Targeted therapy with immunoconjugates for multiple myeloma. Front. Immunol. 2020, 11, 1155.
- U.S. Food and Drug Administration. LUMOXITI (Moxetumomab Pasudotox): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761104s000lbl.pdf (accessed on 20 September 2021).
- AstraZeneca. US FDA Approves Lumoxiti (Moxetumomab Pasudotox-Tdfk) for Certain Patients with Relapsed or Refractory Hairy Cell Leukaemia. Available online: https://www.astrazeneca.com/media-centre/press-releases/2018/us-fda-approves-lumoxiti-moxetumomab-pasudotox-tdfk-for-certain-patients-with-relapsed-or-refractory-hairy-cell-leukaemia.html (accessed on 21 September 2021).
- U.S. Food and Drug Administration. ONTAK (Denileukin Difitox): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/103767s5144lbl.pdf (accessed on 20 September 2021).
- U.S. Food and Drug Administration. ELZONRIS (Tagraxofusp-Erzs): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761116s000lbl.pdf (accessed on 20 September 2021).
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013, 5, 13–21.
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225.
- Russell, M.; Nickerson, D.P.; Odorizzi, G. Molecular mechanisms of late endosome morphology, identity and sorting. Curr. Opin. Cell Biol. 2006, 18, 422–428.
- U.S. Food and Drug Administration. MYLOTARG (Gemtuzumab Ozogamicin): US Prescribing Information 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761060s004lbl.pdf (accessed on 11 August 2021).
- Naito, K.; Takeshita, A.; Shigeno, K.; Nakamura, S.; Fujisawa, S.; Shinjo, K.; Yoshida, H.; Ohnishi, K.; Mori, M.; Terakawa, S.; et al. Calicheamicin-conjugated humanized anti-CD33 monoclonal antibody (gemtuzumab zogamicin, CMA-676) shows cytocidal effect on CD33-positive leukemia cell lines, but is inactive on P-glycoprotein-expressing sublines. Leukemia 2000, 14, 1436–1443.
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496.
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860.
- U.S. Food and Drug Administration. FDA Approves Mylotarg for Treatment of Acute Myeloid Leukemia. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-mylotarg-treatment-acute-myeloid-leukemia (accessed on 1 September 2021).
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: Results of the randomized phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979.
- Taksin, A.-L.; Legrand, O.; Raffoux, E.; De Revel, T.; Thomas, X.; Contentin, N.; Bouabdallah, R.; Pautas, C.; Turlure, P.; Reman, O.; et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: A prospective study of the alfa group. Leukemia 2006, 21, 66–71.
- Renneville, A.; Abdelali, B.R.; Chevret, S.; Nibourel, O.; Cheok, M.; Pautas, C.; Duléry, R.; Boyer, T.; Cayuela, J.-M.; Hayette, S.; et al. Clinical impact of gene mutations and lesions detected by SNP-array karyotyping in acute myeloid leukemia patients in the context of gemtuzumab ozogamicin treatment: Results of the ALFA-0701 trial. Oncotarget 2014, 5, 916–932.
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032.
- Bhattacharya, P.; Basak, A.; Campbell, A.; Alabugin, I.V. Photochemical activation of enediyne warheads: A potential tool for targeted antitumor therapy. Mol. Pharm. 2018, 15, 768–797.
- Vollmar, B.S.; Frantz, C.; Schutten, M.M.; Zhong, F.; del Rosario, G.; Go, M.A.T.; Yu, S.-F.; Leipold, D.D.; Kamath, A.V.; Ng, C.; et al. Calicheamicin antibody–drug conjugates with improved properties. Mol. Cancer Ther. 2021, 20, 1112–1120.
- Hinman, L.M.; Hamann, P.R.; Wallace, R.; Menendez, A.T.; Durr, F.E.; Upeslacis, J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: A novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342.
- Kim, J.H.; Hong, H.J. Humanization by CDR grafting and specificity-determining residue grafting. Antib. Eng. 2012, 907, 237–245.
- Williams, D.G.; Matthews, D.J.; Jones, T. Humanising Antibodies by CDR Grafting; Springer: Berlin/Heidelberg, Germany, 2010; pp. 319–339.
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520.
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of antibody drug conjugates (ADCs). Oncoimmunology 2017, 7, e1395127.
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010.
- Burton, D.R.; Woof, J.M. Human antibody effector function. Adv. Immunol. 1992, 51, 1–84.
- Senter, P.D.; Sievers, E. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637.
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20.
- U.S. Food and Drug Administration. ADCETRIS (Brentuximab Vedotin): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125388s099lbl.pdf (accessed on 20 September 2021).
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; Deblanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465.
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, A.; Picardi, M.; et al. Brentuximab vedotin with chemotherapy for Stage III or IV Hodgkin’s lymphoma. N. Engl. J. Med. 2018, 378, 331–344.
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.I.; Chen, A.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862.
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196.
- Younes, A.; Bartlett, N.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821.
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.; Sussman, D.; Prota, A.E. Structural basis of microtubule destabilization by potent auristatin anti-mitotics. PLoS ONE 2016, 11, e0160890.
- Oflazoglu, E.; Stone, I.J.; Gordon, K.A.; Grewal, I.S.; van Rooijen, N.; Law, C.-L.; Gerber, H.-P. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 2007, 110, 4370–4372.
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2007, 41, 98–107.
- Hughes, B. Antibody–drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667.
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374.
- Oflazoglu, E.; Kissler, K.M.; Sievers, E.; Grewal, I.; Gerber, H.-P. Combination of the anti-CD30-auristatin-E antibody-drug conjugate (SGN-35) with chemotherapy improves antitumour activity in Hodgkin lymphoma. Br. J. Haematol. 2008, 142, 69–73.
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070.
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 2002, 13, 855–869.
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540.
- Kumar, A.; White, J.; Christie, R.J.; Dimasi, N.; Gao, C. Antibody-drug conjugates. Annu. Rep. Med Chem. 2017, 50, 441–480.
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291.
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody–drug conjugates. Chem. Soc. Rev. 2020, 50, 1305–1353.
- Pysz, I.; Jackson, P.J.M.; Thurston, D.E. Introduction to Antibody–Drug Conjugates (ADCs); The Royal Society of Chemistry: London, UK, 2019; Chapter 1; pp. 1–30.
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932.
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2—Positive breast cancer. Clin. Cancer Res. 2010, 16, 4769–4778.
More
Information
Subjects:
Biochemical Research Methods
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.9K
Entry Collection:
Peptides for Health Benefits
Revisions:
2 times
(View History)
Update Date:
16 Nov 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No