The G-protein coupled receptor 55 (GPR55) was first described in 1999 and is broadly expressed in different areas of the CNS, such as the frontal cortex or the hippocampus. The discovery of the bioactive lipid lysophosphtatidylinositol (LPI) as endogenous GPR55 agonist led to the receptor’s deorphanization . However, besides LPI, several commercially available as well as endogenous ligands show agonistic or antagonistic activity at the GPR55. Endocannabinoids, 2-arachidonoylglycerol, and delta-9-tetrahydrocannabinol (Δ9-THC) for instance, show strong affinities and activation of GPR55, heating up the discussion about GPR55 as potential third cannabinoid-receptor (CB). Commercially available GPR55 agonists, such as O-1602, and GPR55-antagonists like ML-193 are commonly used in GPR55 research, to evaluate GPR55-specific molecular pathways and effects. Besides these widely used GPR55 ligands, coumarin-derivates show antagonistic coupled to inverse agonistic activities on GPR55-dependent neuroinflammatory processes as reported recently.
1. Introduction
Oxidative stress has recently gained more attention in the etiology and pathogenesis of neurological and psychiatric diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and depression
[1][2]. Together with neuroinflammation, oxidative stress leads to progressive neurodegeneration, the common path of the mentioned diseases
[3]. However, treatment of neurological and psychiatric diseases still focuses on symptomatic relief without targeting the underlying causal pathological molecular mechanisms. With an aging demography and rising incidences of AD
[4] and PD
[5], adequate and causal treatment with mild side-effects gains even more importance. Therefore, evaluating the role of different receptors and pathways in the homeostasis of oxidative stress in the central nervous system (CNS) might offer new opportunities in the therapy of neurological and psychiatric diseases.
The G-protein coupled receptor 55 (GPR55) was first described in 1999
[6] and is broadly expressed in different areas of the CNS, such as the frontal cortex or the hippocampus
[7]. The discovery of the bioactive lipid lysophosphtatidylinositol (LPI) as endogenous GPR55 agonist led to the receptor’s deorphanization
[8]. However, besides LPI, several commercially available as well as endogenous ligands show agonistic or antagonistic activity at the GPR55
[7][9]. Endocannabinoids, 2-arachidonoylglycerol, and delta-9-tetrahydrocannabinol (Δ9-THC) for instance, show strong affinities and activation of GPR55, heating up the discussion about GPR55 as potential third cannabinoid-receptor (CB)
[7]. Commercially available GPR55 agonists, such as O-1602, and GPR55-antagonists like ML-193 are commonly used in GPR55 research, to evaluate GPR55-specific molecular pathways and effects.
Besides these widely used GPR55 ligands, coumarin-derivates show antagonistic coupled to inverse agonistic activities on GPR55-dependent neuroinflammatory processes as reported recently
[10][11]. Agonistic activities at G-protein coupled receptors (GPCRs) affect downstream signal cascades activated by the receptor, such as the phosphorylation of protein kinases or the translocation of transcriptional factors in the cell’s nucleus. This “On”-state of a receptor can be changed to “Off” spontaneously or by an antagonist, inhibiting competitively or sterically further agonistic binding or spontaneous receptor activation. This “Off”-state of the receptor after binding of an antagonist is equivalent to the receptor’s physiological resting state
[12][13]. Antagonists with inverse agonistic activities, as shown for coumarin derivates, are not just setting a receptor to its resting state, they also induce contrary intracellular signaling as provoked by the receptor’s agonists
[12]. Inverse agonists selectively activate different pathways of the bound receptors, leading to opposite effects in comparison to classical receptor agonists
[14][15]. Therefore, inverse agonism reveals completely new therapeutic options avoiding and reversing pathological receptor activity
[12]. On a molecular level, the inversed agonism is explained by differences in the discriminatory activation of G
α or G
βγ [16] as well as differentiated phosphorylation of GPCRs
[13]. KIT 17, a Coumarin-based compound and GPR55 ligand, showed anti-neuroinflammatory effects in LPS-stimulated primary microglia cells probably relying on an inverse agonistic activity at GPR55 for instance
[10].
The role of GPR55 in the regulation of cellular oxidative homeostasis is still poorly understood. Since GPR55 regulates protein kinase C (PKC), phosphatidyl-inositol-3-kinase (PI3K), and protein kinase B (Akt) in its downstream signaling, GPR55 activation might enforce nuclear factor erythroid 2-related factor 2 (Nrf2) expression
[17][18]. As transcriptional factor, Nrf2 increases the synthesis of antioxidative enzymes and molecules when cells are facing oxidative stress
[18]. However, the activation of GPR55 is connected with increased concentrations of Reactive Oxygen Species (ROS) and apoptosis
[19]. In human aortic endothelial cells, the GPR55 inverse agonist CID16020046 reversed pro-oxidative effects induced by oxidized low-density lipoprotein (LDL) underlining the potential of inverse agonists of GPR55 to interfere with regulatory processes of oxidative stress
[20].
There are multiple assays to evaluate concentrations of ROS and oxidative stress, which are commonly used in research. In this current study, we measured 8-Iso-PGF
2α, a good known and sensitive marker for oxidative stress, to screen for anti-oxidative effects of the coumarin-derivates
[1]. High levels of ROS lead to lipid peroxidation in the cells, and oxidized lipids are increasing levels of isoprostanes
[21]. Besides 8-Iso-PGF
2α-levels, we examined the antioxidative capacity with the oxygen radical antioxidant capacity (ORAC) activity assay, which compares antioxidative properties of compounds to a vitamin E analogue named trolox. The well-established assay measures fluorescent degradation after inducing oxygen radical generation and effects of the tested compounds on prevention of the fluorescent degradation
[22]. Furthermore, we introduce a modified cell viability assay to evaluate prevention of H
2O
2-induced cell death after pretreatment with compounds.
The role of GPR55 in the pathogenesis of different diseases, such as obesity and diabetes, bone disorders, like osteoporosis, and various cancers has been shown before
[9], but the role and possibly therapeutic potential of the GPR55 in CNS disorders such as depression, AD, and PD is still poorly understood
[23][24]. Since the involvement of oxidative stress in the pathogenesis of these diseases is part of actual research, antioxidative GPR55-dependent pathways might be a promising target in future pharmacological therapies.
2. Cytotoxic Effects of KIT C and KIT H
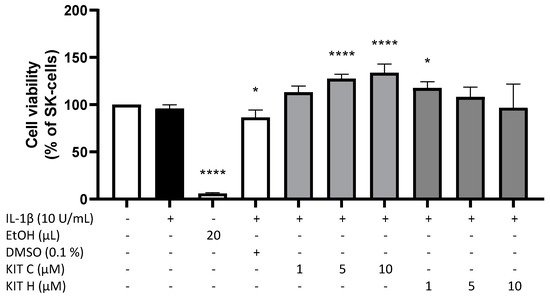
To exclude any cytotoxic effects of the coumarin-based compounds KIT C and KIT H, we performed a MTT-assay to determine the non-toxic concentrations of the compounds. In IL-1β-stimulated SK-N-SH cells, neither KIT C nor KIT H (1–10 µM) significantly decreased the cell viability compared to untreated cells (Figure 1). The solvent of KIT C and KIT H, DMSO, significantly reduced cell metabolism (p < 0.05), whereas 5 and 10 µM of KIT C (p < 0.0001) and 1 µM of KIT H (p < 0.05) significantly increased cell metabolism in the MTT-assay. Since KIT C and KIT H did not reveal cell toxic effect, the tested concentrations up to 10 µM were used for further experiments.
Figure 1. Effects of KIT C (light grey) and KIT H (dark grey) on cell viability of IL-1β-stimulated SK-N-SH cells. Cell viability was measured after 24 h of treatment by change in color due to MTT-oxidation. Values are presented as the mean ± SD of four independent experiments. Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc test with * p < 0.05 and **** p < 0.0001 compared to untreated cells.
3. Effects of KIT C and KIT H on 8-Iso-PGF2α-Release in SK-N-SH and Primary Microglial Cells
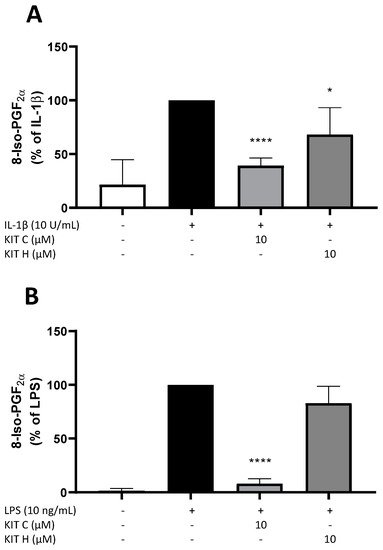
Next, the effects of KIT C and KIT H on the release of 8-Iso-PGF2α in IL-1β-stimulated SK-N-SH cells (Figure 2A) and LPS-stimulated primary mouse microglia cells (Figure 2B) were evaluated using a commercial 8-Iso-PGF2α enzyme immunoassay (EIA). Release of 8-Iso-PGF2α is strongly induced by the stimulation with IL-1β or LPS. In primary microglia cells (Figure 2B), the basal release of 8-Iso-PGF2α was lower compared to SK-N-SH cells (Figure 2A). Ten µM of KIT C significantly reduced 8-Iso-PGF2α-release about 60% in SK-N-SH (p < 0.0001) as well as in microglia cells (90% reduction, p < 0.0001) to levels comparable to the untreated control. The 30% reduction of 8-Iso-PGF2α levels after pre-treatment of SK-N-SH cells with 10 µM KIT H (p < 0.05) was lower compared to KIT C, but still significant compared to IL-1β-stimulated samples. The effects of KIT C and KIT H on 8-Iso-PGF2α levels suggest antioxidative properties of both coumarin-based compounds. Therefore, further experiments were conducted to prove the antioxidative effects and evaluate possible underlying mechanisms.
Figure 2. Effects of KIT C (light grey) and KIT H (dark grey) on the release of 8-Iso-PGF2α in IL-1β-stimulated SK-N-SH cells (A) and LPS-induced primary mouse microglial cells (B). Cells were stimulated as described in material and methods. After 24 h, supernatants were collected and release of 8-Iso-PGF2α was measured by EIA. Values are presented as the mean ± SD of at least three independent experiments. Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc tests with * p < 0.05 and **** p < 0.0001 compared to IL-1β or LPS.
4. Antioxidative Capacity of KIT C and H in the ORAC Assay
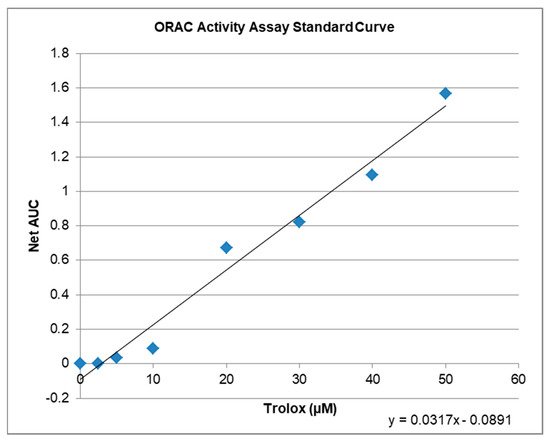
The ORAC-assay determines the antioxidant capacity of compounds ex vitro. The raw values of the tested compounds are compared to a trolox standard curve (Figure 3) and can be calculated as Trolox Equivalents (TE; Table 1). Both, KIT C and KIT H, showed antioxidative capacities in the ORAC assay. The maximal antioxidant capacity was achieved using 10 µM KIT C being comparable to 6.5 µM of trolox. Ten µM of KIT H showed antioxidant capacity comparable to 4.7 µM of Trolox. Interestingly, the antioxidant capacity of KIT C and KIT H is not correlated with the trolox standard curve but seems to reach its plateau at concentrations around 10 µM. The antioxidant capacity of KIT C and KIT H is, therefore, the maximal half of the trolox antioxidative capacity when comparing the concentrations of 10 µM of KIT C and KIT H.
Figure 3. ORAC activity assay standard curve with regression equation y = 0.0317x − 0.0891, that was used to calculate the µM Trolox™ Equivalents (TE) of the tested compounds.
Table 1. ORAC activity assay data for KIT C and KIT H. Raw values and Trolox™ Equivalents in µM.
| Compound |
µM TE 1 |
Net AUC 2 |
AUC |
| Blank |
0.0 |
0.0 |
1.3684 |
| 1 µM KIT C |
5.2 |
0.0743 |
1.4427 |
| 10 µM KIT C |
6.5 |
0.1176 |
1.4860 |
| 1 µM KIT H |
3.4 |
0.0201 |
1.3885 |
| 10 µM KIT H |
4.7 |
0.0607 |
1.4291 |
5. GPR55-Dependent Antioxidative Effects
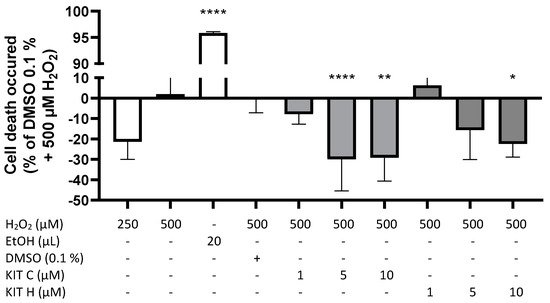
A modified MTT-assay was designed and conducted to evaluate concentration-dependent effects of KIT C and KIT H on ROS-induced cell death in SK-N-SH cells (Figure 4). Figure 4 demonstrates prevention of cell death induced by oxidative stress with negative spikes compared to the baseline. Positive values represent increased cell death. Two hundred fifty µM of H2O2 showed less induction of cell death compared to 500 µM H2O2. Twenty µL of ethanol (approximately 20% EtOH final concentration) reliably induced cell death (p < 0.0001). KIT C concentration-dependent and significantly decreased ROS-induced cell death about maximal 30% with maximal effects using 5 and 10 µM (p < 0.0001 and p < 0.01). KIT H showed less efficient concentration-dependent prevention of ROS-induced cell death compared to KIT C, with a significant reduction at concentrations of 10 µM of KIT H and maximal inhibition of around 23% (p < 0.05).
Figure 4. Effects of KIT C (light grey) and KIT H (dark grey) on H2O2-induced cell death in the ROS-MTT assay in SK-N-SH cells. SK-cells were stimulated as described in material and methods. After 24 h, cell death was measured by change in color due to MTT-oxidation. Values are presented as the mean ± SD of at least three independent experiments. Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc tests with * p < 0.05, ** p < 0.01 and **** p < 0.0001 compared to 500 µM H2O2 with 0.1% DMSO.
 +1 credit
+1 credit