Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Amedeo Amedei | + 2553 word(s) | 2553 | 2021-11-14 12:57:32 | | | |
| 2 | Jason Zhu | Meta information modification | 2553 | 2021-11-16 02:48:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Amedei, A. Gut-Liver Axis and Liver Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/16015 (accessed on 23 July 2026).
Amedei A. Gut-Liver Axis and Liver Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/16015. Accessed July 23, 2026.
Amedei, Amedeo. "Gut-Liver Axis and Liver Diseases" Encyclopedia, https://encyclopedia.pub/entry/16015 (accessed July 23, 2026).
Amedei, A. (2021, November 15). Gut-Liver Axis and Liver Diseases. In Encyclopedia. https://encyclopedia.pub/entry/16015
Amedei, Amedeo. "Gut-Liver Axis and Liver Diseases." Encyclopedia. Web. 15 November, 2021.
Copy Citation
The gut-liver axis has an impact on pathogenesis of numerous chronic liver diseases such as chronic hepatitis B (CHB), chronic hepatitis C (CHC), alcoholic liver disease (ALD), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), development of liver cirrhosis, and hepatocellular carcinoma (HCC).
Gut microbiota
Gut-Liver Axis
Disbyosis
1. Gut-Liver Axis
Gut microbiota (GM) as a “virtual metabolic organ” makes axis with a number of extraintestinal organs, such as kidneys, brain, cardiovascular, and the bone system, but the gut-liver axis attracts increased attention in recent years [1]. The gut-liver axis is a consequence of a close anatomical and functional, bidirectional interaction of the gastrointestinal tract and liver, primarily through a portal circulation. The symbiotic relationship between the GM and the liver is regulated and stabilized by a complex network of interactions, that encompass metabolic, immune, and neuroendocrine crosstalk between them [2]. The tight junctions (TJ) within the gut epithelium represent a natural barrier to bacteria and their metabolic products [3]. Antigens (Ag) (originating either from pathogenic micro-organisms or from food) that pass through these connections, are recognized by dendritic cells, or activate the adaptive immune system by modulating the T cell response. Minimal concentrations of pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides (LPS), peptidoglycans, and flagelin, activate the nuclear factor kappa B (NFKβ) through toll-like receptors (TLRs) and nod-like receptors (NLRs), which leads to the production of inflammatory cytokines and chemokines that enter portal circulation. In addition to hepatocyte damage, PAMPs can activate stellate cells involved in fibrosis promotion and progression, while Kupffer cells are even more responsive to LPS than hepatocytes [4]. Since the gut-liver axis affects the pathogenesis of liver diseases, it is an important focus of current clinical research (Scheme 1).
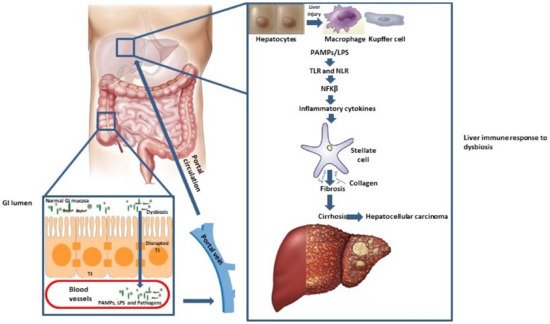
Scheme 1. Gut-liver axis pathogenesis. Abbreviations: GI, gastrointestinal, LPS, Lipopolysaccharides, NFKβ, nuclear factor kappa B, NLR, Nod-like receptors, PAMPs, Pathogen-associated molecular patterns, TJ, Tight junctions, TLR, Toll-like receptors.
2. Disbyosis and Liver Diseases
In general, an increased intestinal permeability and bacterial translocation could enable microbial metabolites to reach the liver, which would impair the bile acid (BA) metabolism and promote gut dysmotility and systemic inflammation. All these conditions could induce gut dysbiosis, which, in turn, further increases liver damage. It has been observed that the stage of liver injury correlates closely with the severity of gut dysbiosis [5]. Alterations in the fecal bacterial flora are described by changes in the composition of the dominant Bacteroidetes and Firmicutes phyla, including Ruminococcaceae, Lachnospiraceae, and Clostridiales, which produce short-chain fatty acids (SCFA) that are an energy source for the intestinal epithelium’s cells, but can also regulate secondary BA metabolism and induce a regulatory immune process and IgA production.
2.1. Hepatitis B Virus (HBV) Infection
CHB is an important health issue worldwide. Acute HBV infection leads to CHB in just 5% of adult patients, while the proportion is quite different in children, since more than 90% of exposed neonates and 30% to 50% of children aged 1 to 5 years fail in HBV clearance. Liver injury is mediated by HBV induced immune response. TLRs play an important role in the production of interferons and proinflammatory cytokines and immune cells recruitment in order to suppress viral replication. It has been established that age-specific seroclearance depends not only on the maturity of the immune system, but on the GM stability as well [6]. Involvement of GM in HBV clearance was demonstrated in animal models. Chou et al. showed that adult mice with mature GM managed to clear HBV after six weeks of infection, which is the opposite among young mice without GM, who remained HBV positive. The fact that adult mice failed to clear HBV after gut sterilization by antibiotics (6 to 12 weeks), emphasizes the GM significance in anti-HBV immunity [7]. It also implies new therapeutic strategy for patients with HBV infection [8]. In fact, the transplantation of fecal microbiota (FMT), in addition to standard antivirals, has been shown to be effective in HBeAg clearance [9].
Compositional and structural changes of GM have been detected in patients with CHB and liver cirrhosis. These patients have a decreased ratio of Bifidobacteriaceae/Enterobacteriaceae (B/E), based on low levels of Bifidobacteria and Lactobacillus, and high levels of Enterococcus and Enterobacteriacea. In addition, gut permeability is altered when accompanied with bacterial translocation and the presence of endotoxins in the portal vein, which leads to increased TLS/NLR activation in the liver with consequential cytokine production and occurrence of liver lesions, progression of fibrosis, and development of cirrhosis and HCC [10][11]. Wei et al. have demonstrated that GM of patients with HBV-related cirrhosis contained lower levels of Bacteroidetes (4% vs. 53%) and increased levels of Proteobacteria (43% vs. 4%) compared to the heathy group [12]. In an other study, patients with alcohol-related and HBV-related cirrhosis showed decreased GM diversity, compared to healthy individuals, with a predominance of Enterobacteriaceae and Streptococcaceae [13].
2.2. Hepatitis C Virus (HCV) Infection
CHC is a global health problem that leads to progressive liver fibrosis and the cirrhosis development in 20% to 30% of untreated patients after 20 to 30 years. It has been estimated that 1% to 4% of these patients develop HCC each year [14]. GM has been rarely analyzed in patients with HCV infection. According to published data, the GM found in HCV patients shows lower microbial diversity in comparison to those in healthy controls [15][13][16]. CHC could alter microbiota composition through IgA produced by HCV infected gastric B-lymphocyte. GM found in Egyptian patients with CHC contains more Prevotella and Faecalibacterium and less Acinetobacter, Veillonella, and Phascolarctobacterium than healthy individuals. In the study of Aly et al., Bifidobacterium was detected only in GM of the healthy group, posing the possible new role of Prevotella/Faecalibacterium vs. Bifidobacterium ratio as a biomarker for CHC and fibrosis progression [15]. Disease progression could bring more profound changes in CHC patients’ GM. Therefore, according to Heidrich et al., decreased diversity was more pronounced in HCV patients’ with established cirrhosis than in those with less advanced CHC [17]. Liver cirrhosis per se could be an independent risk factor for dysbiosis regardless of the HCV viral load. This hypothesis is in agreement with a study performed by Bajaj et al. who found that patients with HCV cirrhosis have gut dysbiosis regardless of long-term HCV eradication. A sustained virological response (SVR) did not improve gut dysbiosis in patients with HCV cirrhosis, due to refractory systemic inflammation and endotoxemia in these individuals [18]. Bacterial translocation was described in patients with CHC and together with increased intestinal permeability (“leaky gut”), it poses a well-established milieu with TLR/NLR activation and expression of pro-inflammatory cytokine genes, especially in those with cirrhosis [19].
2.3. Alcoholic Liver Disease
Alcohol abuse is a prominent cause of liver damage worldwide. GM is recognized as a key player in the severity of liver injury in ALD, in addition to the quantity of consumed alcohol and genetic predisposition (patatin like phospholipase domain containing 3 (PNPLA3), Transmembrane 6 superfamily 2 human gene (TM6SF2), membrane bound O-acyltransferase domain containing 7 (MBOAT7), and solute carrier family 38 member 4 (SLC38A4) etc.) [20]. Alcohol consupmtion leads to small and large intestine overgrowth and modulation of GM composition in both animals and humans [21]. Alcohol and its degradation products disrupt epithelial TJ leading to increased intestinal permeability and inflammation [20]. Gut-derived PAMPs (e.g., endotoxin) are increased after heavy alcohol intake [22]. Ethanol consumption alters the GM composition through SCFAs modulation. Intestinal levels of SCFAs are lower after alcohol consumption with the exception of increases in acetic acid levels, which is the metabolite of ethanol [23]. Alcoholic abuse was shown to be associated with decreased levels of butyrate-producing Clostridiales species order and increased levels of pro-inflammatory Enterobacteriaceae. In those with established cirrhosis, multiple members of the Bacteroidales order were depleted with a rise of taxa normally inhabiting the oral cavity [24].
2.4. Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis
NAFLD is one of the most important causes of liver disease worldwide, with global prevalence of 25%. NAFLD is one of the top risk factors for HCC and is predicted to become the most common indication for liver transplantation [25]. NAFLD is a consequence of triglyceride accumulation in the hepatocytes and is considered to be the hepatic manifestation of obesity and the metabolic syndrome [26]. About 20% of patients with NAFLD develop NASH, which is a chronic hepatic inflammation that can progress to cirrhosis, end stage liver disease (ESLD), and HCC. Pathogenesis of NASH is not yet fully elucidated, but it is described as a “two hit” phenomenon. The primary event is lipid accumulation with alterations of lipid homeostasis associated with obesity, insulin resistance, and adipokine abnormalities. The second “hit” is a combination of oxidative stress, lipid peroxidation, mitochondrial dysfunction, BA toxicity, cytokine-mediated recruitment, and retention of inflammatory cells [27]. Obesity is associated with dysbiotic gut microbiota, with decreased diversity and an increased Firmicutes/Bacteroidetes ratio [28]. A similar Firmicutes/Bacteroidetes ratio was found in diabetes patients as well. Endogenous ethanol is constantly produced by microbiota, regardless of oral alcohol intake, especially in those with a sugar-rich diet. Increased ethanol production by microbiota in obese humans and mice leads to the activation of TLRs in the liver, cytokine production, and alters the BA profile. Endogenous ethanol serum concentrations are significantly higher in patients with NASH compared to obese or healthy controls [29]. Gut dysbiosis in patients with NAFLD/NASH promotes insulin resistance, de novo lipogenesis in liver, and also increases intestinal permeability, which promotes chronic PAMPs exposure and oxidative stress caused by increased endogenous ethanol [30]. Endotoxin/TLR4 signalling contributes to the development of fibrosis and progression to cirrhosis through hepatic stellate cell activation. The GM plays a critical role in the conservation of the mainstream BA pool, which transforms BA to several metabolites by oxidation/epimerization, deconjugation, esterification, 7-dehydroxylation, and desulfatation. Changes in any of these modulations is a cause of disease. GM dysbiosis in NAFLD could affect the conversion of primary bile acids into secondary bile acids [31]. It has been observed that the bacteria able to make this transformation are decreased in the NAFLD cirrhosis fecal samples. In particular, there is a higher level of Enterobacteriaceae (that could be potentially pathogenic) with lower Ruminococcaceae, Lachnospiraceae, and Blautia (with a 7α-dehydroxylating activity) abundances.
Currently, the roles of BA and nuclear receptors are also strongly considered. The BA primarly synthesized in the liver are secreted to the gallbladder and then released into the duodenum following food ingestion. In the gut, the size and composition of the BA pool can be modified by GM via the biotransformation of primary into secondary BA. The BA contributes to the emulsification and fat solubilization but also activates the expression of a nuclear bile acid receptor FXR (farnesoid X receptor) and a membrane G protein-coupled receptor TGR5. The reduction of the secondary BA synthesis attributed to GM dysbiosis lowers the activation of nuclear receptors FXR and TGR5 in the ileum, which leads to retained bile salts, perpetuation of gut permeability, small bowel translocation, and bacterial overgrowth, which contributes to liver disease [32]. The FXR plays a key role in mediating the crosstalk between the host and GM, especially through the modulation of enterohepatic BA circulation. FXR exerts bile-acid regulatory effects via a tissue-specific mechanism. [33]. In detail, in the liver, FXR induces the expression of the small heterodimer partner (SHP), which inhibits CYP7A1 (Cholesterol 7 alpha-hydroxylase) expression, while, in the intestine, FXR increases the levels of circulating fibroblast growth factor 19 (FGF19), which decrease the expression of CYP7A1 and cytochrome P450 12a-hydroxylase B1 (CYP8B1). Therefore, this leads to the inhibition of BA synthesis [34].
FXR activation has been known to reduce triglyceride levels suppressing the synthesis and uptake of the fatty acids in the liver [35]. In addition, the FXR roles in decreasing inflammation have been emerging [36]. Lastly, related to glucide metabolism, FXR reduces insulin resistance, gluconeogenesis, increases glycogenesis, and, therefore, decreases the blood glucose amount. A more fine elucidation of FXR functions in liver and intestine needs further research. Furthermore, mice models showed that FXR activation, induced by BA products (converted by GM) might protect against bacterial overgrowth, gut permeability, and small bowel translocation [37]. The degree of FXR activation could be regulated by GM dysbiosis, inducing BA alteration and, hence, liver disease secondary to retained bile salts and a leaky gut. Bacteria, translocated from the gut, may additionally decrease FXR activation within hepatocytes, which leads to decreased BSEP (bile salt export pum) activity.
The bile acid-activated FXR and TGR5 has a protective role in liver disease progression, which means the activation of both receptors has been proposed as therapy. Many activators of FXR and TGR5 have been developed from bile acid analogues, which are able to decrease hepatic steatosis and inflammation [38]. FXR agonists such as obeticholic acid have already been recognized as a new therapeutic venue for NASH and cholestatic diseases. NASH patients exhibit increased fecal primary BAs, primary/secondary BAs fecal ratio, and plasma and hepatic BAs concentrations [39]. This cytotoxicity can lead to NASH progression and finally to liver cirrhosis.
2.5. Hepatic Encephalopathy (HE) and Spontaneous Bacterial Peritonitis (SBP)
In all patients with ESLD, regardless of its etiology, HE and SBP commonly occur. HE is considered to be a consequence of high ammonia level, but GM and bacteria’s products, such as amino acid metabolites (indoles, oxindoles) and endotoxins, are also involved in HE development. The connection between HE and GM is the production of ammonia and endotoxins by urease-producing bacteria, such as Klebsiella and Proteus, which are present in GM [40]. It has been shown that microbiota of the sigmoid colon in liver cirrhosis patients differs in those with HE when compared with patients without HE [41]. Gut dysbiosis in liver cirrhosis also contributes to the development of Spontaneous Bacterial Peritonitis (SBP) through damaged intestinal barrier and a higher degree of microbial translocation [42].
2.6. Hepatocellular Carcinoma
HCC is the most common primary malignancy in adults with chronic liver disease and liver cirrhosis [43]. HBV and HCV infection, alcohol abuse, and dietary aflatoxin are major risk factors for the development of HCC. Although HBV and HCV account for 80% to 90% of overall HCC, regarding the obesity pandemic, an emergance of potent direct acting antivirals for HCV and worldwide available vaccine for HBV, it can be expected that HCC epidemiology will change in the future [44]. An alarming rise in the incidence of NAFLD and NASH was accompanied by an increased development of NASH-related HCC incidence [45].
All previously mentioned mechanisms, leaky gut, endotoxemia, TLR, dysbiosis, and immunomodulation promote the development of HCC [46]. The gastrointestinal tract contributes to homeostasis by maintaining an intact barrier against LPS and intestinal bacteria. In the case of increased intestinal permeability, bacterial translocation and LPS accumulation will lead to intestinal bacterial overgrowth and changes of GM composition. In patients with chronic liver diseases/cirrhosis, detoxification, degradation, and clearance of LPS and other bacterial products is compromised [47]. Altered microbiota is generally presented in HCC patients [48]. HCC patients have been reported to have high levels of Escherichia coli and other gram-negative bacteria in GM, which are associated to increased LPS serum levels [49]. Oribacterium and Fusobacterium are the most commonly isolated bacteria from tongue swab of HCC patients. On the other side, GM of HCC patients contains reduced levels of Lactobacillus spp., Bifidobacterium spp., and Enterococcus spp. [50]. Unlike merely microbial species, it was shown that microbial metabolism, iron transport, and energy-producing system significantly differ between GM of HCC patients and healthy control [48]. TLR4 expressed by activated stellate cells react to low LPS concentrations, which ensures the development of fibrosis and cirrhosis. The significance of GM and TLR4 activation in hepatocarcinogenesis have been studied in an animal model. It has been demonstrated that GM and TLR4 activation promote HCC development by increased cell proliferation and suppression of apoptosis [51].
References
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut–Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. (Basel) 2018, 6, 79.
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835.
- Vajro, P.; Paolella, G.; Fasano, A. Microbiota and gut-liver axis: A mini-review on their influences on obesity and obesity-related liver disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 461–468.
- Yiu, J.H.; Dorweiler, B.; Woo, C.W. Interaction between gut microbiota and toll-like receptor: From immunity to metabolism. J. Mol. Med. 2017, 95, 13–20.
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947.
- Yang, R.; Xu, Y.; Dai, Z.; Lin, X.; Wang, H. The Immunologic Role of Gut Microbiota in Patients with Chronic HBV Infection. J. Immunol. Res. 2018, 2018, 2361963.
- Chou, H.H.; Chien, W.H.; Wu, L.L.; Cheng, C.H.; Chung, C.H.; Horng, J.H.; Ni, Y.H.; Tseng, H.T.; Wu, D.; Lu, X.; et al. Age-related immune clearance of hepatitis B virus infection requires the establishment of gut microbiota. Proc. Natl. Acad. Sci. USA 2015, 112, 2175–2180.
- Kang, Y.; Cai, Y. Gut microbiota and hepatitis-B-virus-induced chronic liver disease: Implications for faecal microbiota transplantation therapy. J. Hosp. Infect. 2017, 96, 342–348.
- Ren, Y.D.; Ye, Z.S.; Yang, L.Z.; Jin, L.X.; Wei, W.J.; Deng, Y.Y.; Chen, X.X.; Xiao, C.X.; Yu, X.F.; Xu, H.Z.; et al. Fecal microbiota transplantation induces hepatitis B virus e-antigen (HBeAg)clearance in patients with positive HBeAg after long-term antiviral therapy. Hepatology 2017, 65, 1765–1768.
- Chassaing, B.; Etienne-Mesmin, L.; Gewirtz, A.T. Microbiota-liver axis in hepatic disease. Hepatology 2014, 59, 328–339.
- Wang, J.; Wang, Y.; Zhang, X.; Liu, J.; Zhang, Q.; Zhao, Y.; Peng, J.; Feng, Q.; Dai, J.; Sun, S.; et al. Gut Microbial Dysbiosis Is Associated with Altered Hepatic Functions and Serum Metabolites in Chronic Hepatitis B Patients. Front. Microbiol. 2017, 8, 2222.
- Wei, X.; Yan, X.; Zou, D.; Yang, Z.; Wang, X.; Liu, W.; Wang, S.; Li, X.; Han, J.; Huang, L.; et al. Abnormal fecal microbiota community and functions in patients with hepatitis B liver cirrhosis as revealed by a metagenomic approach. BMC Gastroenterol. 2013, 13, 175.
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572.
- Lee, M.H.; Yang, H.I.; Yuan, Y.; L’Italien, G.; Chen, C.J. Epidemiology and natural history of hepatitis C virus infection. World J. Gastroenterol. 2017, 20, 9270–9280.
- Aly, A.M.; Adel, A.; El-Gendy, A.O.; Essam, T.M.; Aziz, R.K. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. 2016, 8, 42.
- Preveden, T.; Scarpellini, E.; Milić, N.; Luzza, F.; Abenavoli, L. Gut microbiota changes and chronic hepatitis C virus infection. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 813–819.
- Heidrich, B.; Vital, M.; Plumeier, I.; Döscher, N.; Kahl, S.; Kirschner, J.; Ziegert, S.; Solbach, P.; Lenzen, H.; Potthoff, A.; et al. Intestinal microbiota in patients with chronic hepatitis C with and without cirrhosis compared with healthy controls. Liver Int. 2018, 38, 50–58.
- Bajaj, J.S.; Sterling, R.K.; Betrapally, N.S.; Nixon, D.E.; Fuchs, M.; Daita, K.; Heuman, D.M.; Sikaroodi, M.; Hylemon, P.B.; White, M.B.; et al. HCV eradication does not impact gut dysbiosis or systemic inflammation in cirrhotic patients. Aliment. Pharmacol. Ther. 2016, 44, 638–643.
- Munteanu, D.; Negru, A.; Radulescu, M.; Mihailescu, R.; Arama, S.S.; Arama, V. Evaluation of bacterial translocation in patients with chronic HCV infection. Rom. J. Intern. Med. 2014, 52, 91–96.
- Cassard, A.M.; Ciocan, D. Microbiota, a key player in alcoholic liver disease. Clin. Mol. Hepatol. 2017, 24, 100–107.
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol Clin. Exp. Res. 2015, 39, 763–775.
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2014, 148, 30–36.
- Xie, G.; Zhong, W.; Zheng, X.; Li, Q.; Qiu, Y.; Li, H.; Chen, H.; Zhou, Z.; Jia, W. Chronic ethanol consumption alters mammalian gastrointestinal content metabolites. J. Proteome Res. 2013, 12, 3297–3306.
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 141.
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2018, 30335697.
- Boppidi, H.; Daram, S.R. Nonalcoholic fatty liver disease: Hepatic manifestation of obesity and the metabolic syndrome. Postgrad. Med. 2008, 120, 1–7.
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 981–992.
- Chakraborti, C.K. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119.
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609.
- Poeta, M.; Pierri, L.; Vajro, P. Gut-Liver Axis Derangement in Non-Alcoholic Fatty Liver Disease. Children 2017, 4, 66.
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955.
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744.
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell. 2000, 6, 517–526.
- Leung, D.H.; Yimlamai, D. The intestinal microbiome and paediatric liver disease. Lancet Gastroenterol. Hepatol. 2017, 2, 446–455.
- Zhu, Y.; Li, F.; Guo, G.L. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol. Res. 2011, 63, 259–265.
- Shaik, F.B.; Prasad, D.V.; Narala, V.R. Role of farnesoid X receptor in inflammation and resolution. Inflamm. Res. 2015, 64, 9–20.
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925.
- Oliveira, M.C.; Gilglioni, E.H.; Boer, B.A.; Waart, D.R.; Salgueiro, C.L.; Ishii-Iwamoto, E.L.; Oude Elferink, R.P.J.; Gaemers, I.C. Bile acid receptor agonists INT747 and INT777 decrease oestrogen deficiency-related postmenopausal obesity and hepatic steatosis in mice. Biochim. Biophys. Acta 2016, 1862, 2054–2062.
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile acid control of metabolism and iInflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 2017, 152, 1679–1694.
- Häussinger, D.; Schliess, F. Pathogenetic mechanisms of hepatic encephalopathy. Gut 2008, 57, 1156–1165.
- Rai, R.; Saraswat, V.A.; Dhiman, R.K. Gut microbiota: Its role in hepatic encephalopathy. J. Clin. Exp. Hepatol. 2015, 5, 29–36.
- Oikonomou, T.; Papatheodoridis, G.V.; Samarkos, M.; Goulis, I.; Cholongitas, E. Clinical impact of microbiome in patients with decompensated cirrhosis. World J. Gastroenterol. 2018, 24, 3813–3820.
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2018. S0016-5085(18)35165-5.
- Nordenstedt, H.; White, D.L.; El-Serag, H.B. The changing pattern of epidemiology in hepatocellular carcinoma. Dig. Liver Dis. 2010, 42, 206–214.
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543.
- Wan, M.L.Y.; El-Nezami, H. Targeting gut microbiota in hepatocellular carcinoma: Probiotics as a novel therapy. Hepatobiliary Surg. Nutr. 2018, 7, 11–20.
- Tao, X.; Wang, N.; Qin, W. Gut Microbiota and Hepatocellular Carcinoma. Gastrointest. Tumors. 2015, 2, 33–40.
- Lu, H.; Ren, Z.; Li, A.; Zhang, H.; Jiang, J.; Xu, S.; Luo, Q.; Zhou, K.; Sun, X.; Zheng, S.; et al. Deep sequencing reveals microbiota dysbiosis of tongue coat in patients with liver carcinoma. Sci. Rep. 2016, 6, 33142.
- Grat, M.; Wronka, K.M.; Krasnodebski, M.; Masior, L.; Lewandowski, Z.; Kosinska, I.; Grat, K.; Stypulkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of gut microbiota associated with the presence of hepatocel- lular cancer in patients with liver cirrhosis. Transplant Proc. 2016, 48, 1687–1691.
- Zhang, H.L.; Yu, L.X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.X.; Shan, L.; Liu, Q.; et al. Profound impact of gut homeostasis on chemically induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812.
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012, 21, 504–516.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
16 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No