Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gianluigi Mazzoccoli | + 2950 word(s) | 2950 | 2021-11-04 07:15:26 | | | |
| 2 | Catherine Yang | Meta information modification | 2950 | 2021-11-15 09:39:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mazzoccoli, G. Left Ventricular Hypertrophy Pathophysiology. Encyclopedia. Available online: https://encyclopedia.pub/entry/15989 (accessed on 23 July 2026).
Mazzoccoli G. Left Ventricular Hypertrophy Pathophysiology. Encyclopedia. Available at: https://encyclopedia.pub/entry/15989. Accessed July 23, 2026.
Mazzoccoli, Gianluigi. "Left Ventricular Hypertrophy Pathophysiology" Encyclopedia, https://encyclopedia.pub/entry/15989 (accessed July 23, 2026).
Mazzoccoli, G. (2021, November 15). Left Ventricular Hypertrophy Pathophysiology. In Encyclopedia. https://encyclopedia.pub/entry/15989
Mazzoccoli, Gianluigi. "Left Ventricular Hypertrophy Pathophysiology." Encyclopedia. Web. 15 November, 2021.
Copy Citation
Left ventricular hypertrophy (LVH) can be adaptive, as arising from exercise, or pathological, most commonly when driven by hypertension. The pathophysiology of LVH is consistently associated with an increase in cytochrome P450 (CYP)1B1 and mitogen-activated protein kinases (MAPKs) and a decrease in sirtuins and mitochondria functioning. The pathoetiology of LVH is intimately associated with increased blood pressure and therefore with the array of different factors associated with hypertension, including the various manifestations and consequences of stress, obesity and diabetes.
left ventricular hypertrophy
1. Mitogen-Activated Protein Kinase (MAPK) Pathways
Numerous intracellular pathways have been associated with LVH, as with many proliferative conditions [1], including activation of the MAPK pathways, which are strongly associated with both exercise-induced and pathophysiological cardiac hypertrophy [2][3][4][5][6]. The MAPK pathways are important regulators of many physiological and pathophysiological processes, with roles in cell proliferation and stress responses. The protein 38 (p38) MAPK branch of the MAPK pathways can be activated under pressure load, including hypertension [4]. A wide array of studies using different experimental paradigms show pathological cardiac hypertrophy to be significantly attenuated by p38MAPK pathway inhibition [4][5][6]. p38MAPK pathway activity leads to the downstream activation of cyclic adenosine 3′,5′-monophosphate (cAMP)-response element binding protein (CREB), with CREB interacting with DNA and regulating a plethora of gene transcriptions strongly associated with LVH [7]. Brain-derived neurotrophic factor (BDNF) is also increased in LVH [8] and can contribute to LVH pathophysiology via two-way activation interactions with CREB, with increased BDNF also being induced by p38MAPK [9].
A number of studies indicate a role for Src homology-2 domain-containing phosphatase2 (Shp2) in cardiac hypertrophy [10]. Shp2 is an ERK signalling mediator, with Shp2 interactions with tyrosine receptor kinase (Trk)B being necessary for BDNF to activate extracellular signal-regulated kinases (ERK) signalling [11]. It is via the ERK activation pathways and the small GTPases, RhoA and Rac1, that Shp2 acts to regulate the cellular morphology changes occurring in LVH [10]. Shp2 can then act to regulate TrkB-linked MAPK pathway activation by BDNF and other TrkB ligands, including N-acetylserotonin (NAS) [12]. NAS is the immediate precursor of melatonin in the melatonergic pathway (see Figure 1).
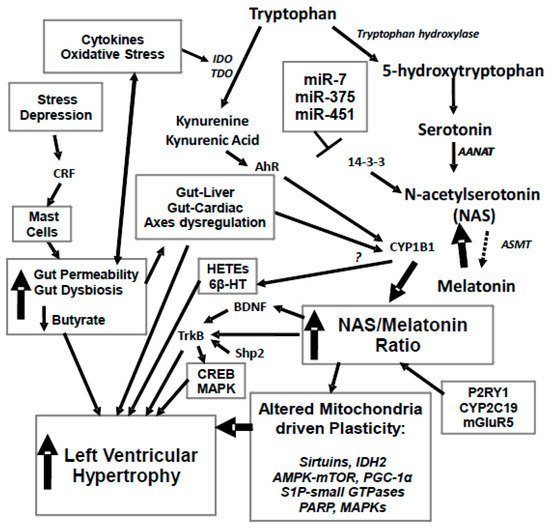
Figure 1. Stress and depression drive an increase in corticotropin releasing factor (CRF) that activates mucosal mast cells and leads to an increase in gut permeability, coupled to gut dysbiosis and decreased butyrate production. This drives alterations in the gut-liver and gut-cardiac axes, with a decrease in butyrate’s histone deacetylase (HDAC) inhibitory activity regulating CYP1B1. Increased gut permeability also drives an increase in oxidative stress and pro-inflammatory cytokines, which activate tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO), leading to a driving of tryptophan down the kynurenine pathway, with a resultant decrease in serotonin, N-acetylserotonin (NAS) and melatonin synthesis. An increase in the NAS/melatonin ratio from CYP1B1 and probably mGluRgpI, P2Y1 receptor, CYP2C19 and O-demethylation leads to NAS activation of TrkB, as well as brain-derived neurotropic factor (BDNF) induction, in turn increasing cyclic adenosine 3′,5′-monophosphate (cAMP)-response element binding protein (CREB) and the mitogen-activated protein kinase (MAPK) pathways, which all contribute to left ventricular hypertrophy (LVH). The suppression of melatonin drives alterations in mitochondria, including via melatonin’s interactions with the sirtuins and PGC-1α, with consequences for IDH2 and AMPK-mTOR regulation of mitochondria. Alterations in the gut drive changes in the gut-liver and gut-cardiac axes that, along with kynurenine activation of the AhR raises CYP1B1 levels. CYP1B1 may also contribute to LVH via the hydroxyeicosatetraenoic acids (HETEs) and 6β-HT. A number of microRNAs known to be altered in LVH, including miR-7, miR-375 and miR-451, all act to regulate 14-3-3 and therefore the stabilization of AANAT at the start of the melatonergic pathway. Similar processes can underpin hypertension, which is the major driver of LVH. 6β-HT: 6β-hydroxytestosterone; AANAT: aralkylamine N-acetyltransferase; AhR; aryl hydrocarbon receptor; AMPK: AMP-activated protein kinase; ASMT: acetylserotonin O-methyltransferase; BDNF: brain derived neurotrophic factor; CREB: cyclic adenosine 3′,5′-monophosphate (cAMP)-response element binding protein; CRF: corticotrophin releasing factor; CYP: Cytochrome P450; E2: estrogen; GLUT1: glucose transporter 1; HDAC: histone deacetylase; HETEs: hydroxyeicosatetraenoic acids; IDO: indoleamine 2,3-dioxygenase; KAT: kynurenine aminotransferase; LVH: left ventricular hypertrophy; MAPK: mitogen activated protein kinases; mGluR: metabotropic glutamate receptor; Mito: mitochondria; NAS: N-acetylserotonin; P2Y1: purinergic receptor; PARP: poly-(ADP-ribose) polymerase; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; S1P: sphingosine-1-phosphate; TDO: tryptophan 2,3-dioxygenase; TrkB: tyrosine receptor kinase B. Arrows indicate activation; T-bar indicates inhibition.
Overall, the MAPK pathways, including p38 MAPK and the ERKs, are strongly associated with cardiac hypertrophy. Notably, MAPK pathway activation can be driven by raised levels of CYP1B1 [13].
2. CYP1B1
Increased CYP1B1 is evident in cardiac hypertrophy, including when induced by dasatinib [14]. The increase in CYP1B1 may not necessarily be mediated via its classical induction by the AhR [14]. Rather its relevance to cardiac hypertrophy has been attributed to the mid-chain hydroxyeicosatetraenoic acids (HETEs) [13]. These authors showed that the inhibition of CYP1B1 significantly attenuates isoprenol-induced cardiac hypertrophy, with protection afforded by the modulation of superoxide anion, MAPKs, and nuclear factor-κB (NF-κB) [13], whilst the overexpression of CYP1B1 significantly induces cellular hypertrophy and mid-chain HETE metabolites. Other CYP1B1 metabolites have been shown to contribute to cardiac hypertrophy, including 6β-hydroxytestosterone [15].
Increased CYP1B1 is also relevant to the hypertensive pathoetiology of LVH, with CYP1B1 inhibition attenuating the blood pressure increase in spontaneously hypertensive rats (SHR), but not in control rodents, as well as dramatically limiting increases in vascular reactivity, cardiovascular hypertrophy, endothelial dysfunction and renal dysfunction, as well as cardiac and renal fibrosis in SHR [16]. CYP1B1 inhibition also significantly attenuates ROS production and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity in SHR, as well as the heightened plasma levels of pro-inflammatory cytokines and catecholamines [16]. These authors also showed CYP1B1 inhibition to modulate the cardiac activity of p38 MAPK, ERK, c-Src tyrosine kinase, and Akt in SHR [16]. Such data clearly highlight a role for CYP1B1 inhibition in the management of hypertension and its associated disorders, including LVH. As such, raised CYP1B1 levels would seem relevant to both the pathoetiology and pathophysiology of LVH. CYP1B1 also ‘backward’ converts melatonin to NAS, thereby increasing TrkB activation, BDNF, CREB and the MAPKs (Figure 1). CYP1B1 levels are most commonly raised following AhR activity.
3. Aryl Hydrocarbon Receptor (AhR)
AhR activation can drive hypertrophic effects in the cardiomyocyte cell line, H9c2, concurrent to its induction of CYP1B1 in these cells [17]. Work in another cardiomyocyte cell line, AC 16 cells, shows AhR ligands to gradually raise the mRNA and protein levels of the AhR and CYP1(A1/B1), in association with attenuated mitochondrial activity, altered mitochondria membranes and heightened mitochondrial ROS levels [18]. AhR activation is associated with the development of hypertension and LVH [19]. Such data clearly shows the relevance of the AhR and CYP1B1 to the cellular and mitochondria changes occurring in LVH. The AhR induces its own repressor, the AhRR. However, not all genes activated by the AhR are inactivated by the AhRR [20], indicating that AhR activation can have longer term consequences for gene expression patterning.
It should be noted that the AhR has regulatory physiological as well as pathophysiological roles. The AhR can be activated by endogenous ligands, as well as by induced and exogenous ligands [21]. The induction of the kynurenine pathway can increase levels of kynurenine and kynurenic acid, although most work has investigated these changes in preclinical models. Both kynurenine and kynurenic acid activate the AhR and therefore CYP1B1. Heightened activation of the kynurenine pathway is evident in LVH [22]. The inclusion of the kynurenine pathways in the pathophysiology of LVH, readily allows for the inclusion of stress and affective dysregulation in the etiology of both hypertension and LVH (see Figure 1).
4. Oxidative Stress, Immune-Inflammation and Kynurenine Pathways
Heightened levels of oxidative stress and immune-inflammatory activity are evident in LVH, both systemically in clinical samples [23] and in preclinical cardiomyocytes [24], as well as in hypertension patients [25]. Consequently, the induction of oxidative stress is a preclinical model of cardiac hypertrophy [26]. Pro-inflammatory cytokines are similarly increased in hypertension [27] as well as in LVH serum and cardiomyocytes [28][29]. Low-grade inflammation, as indicated by C-reactive protein, and kynurenine pathway activation are linked to adverse cardiac remodelling [22], with raised kynurenine levels long appreciated to modulate the efficacy of antihypertensives [30]. Such systemic and local markers of oxidative stress and immune inflammatory activity are therefore likely to indicate AhR and CYP1B1 activation, as shown in preclinical models, as well as modulate the effects of medications.
The heightened levels of pro-inflammatory cytokines and oxidative stress/ROS in LVH increases the activation of indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO), respectively [31]. IDO and TDO induction drives tryptophan along the kynurenine pathway and away from serotonin and melatonin synthesis [32]. Interleukin (IL)-1B, IL-6, IL-18, tumor necrosis factor (TNF)-α and interferon (IFN)-γ are increased in LVH and hypertension [33], with all these pro-inflammatory cytokines activating IDO. Likewise, oxidative stress, ROS and pro-inflammatory cytokines can induce TDO. Although IDO can regulate blood pressure [34], it has been relatively little investigated in cardiac hypertrophy. This is surprising given the increase in both IDO inducers and kynurenine pathway products in hypertension and cardiovascular disorders more widely. TDO is predominantly expressed in the liver and brain, with effects therefore more likely to be indirect [35]. This is of some importance to the role of the AhR and CYP1B1 in cardiac hypertrophy, as both kynurenine and kynurenic acid activate the AhR to increase CYP1B1.
5. Gut Dysbiosis and Gut Permeability
A plethora of recent studies have highlighted a role for alterations in the gut microbiome, in association with increased gut permeability, in driving hypertension, cardiovascular disorders, and metabolic syndrome more widely [36]. The association of psychological stress with hypertension and LVH [37] may be mediated by stress-induced corticotropin releasing factor (CRF), which acts on mucosal mast cells to increase gut permeability [38]. Gut dysbiosis and increased gut permeability are associated with the transfer of gut bacteria or tiny fragments of partially digested food over the gut barrier, leading to raised levels of circulating lipopolysaccharide (LPS), that acts on the immune system to heighten pro-inflammatory cytokines and ROS [39]. LPS activation of the toll-like receptor (TLR)4 is sufficient to drive LVH [40]. The association of chronic and repetitive stress with anxiety and depression seems at least partially mediated by gut dysbiosis and increased gut permeability [41]. Alterations in gut regulation are therefore an important aspect of how stress and affect dysregulation link to hypertension and LVH [42]. Under conditions of gut dysbiosis, there is a decrease in the microbiome-derived short-chain fatty acid, butyrate. This leads to the loss of butyrate’s protective effects on the maintenance of the gut barrier as well as on the optimization of systemic mitochondria functioning and immune dampening [39]. The latter effects are mediated by the crossing of butyrate over the gut barrier, with butyrate having optimizing effects on mitochondria regulation, at least in part via its induction of melatonin [43].
Clearly, gut dysbiosis and the gut-liver axis are important in mediating the pathophysiological processes associated with type II diabetes and the association of this condition with LVH. However, as well as dietary driven changes in the gut microbiome, the effects of psychological stress and affective dysregulation can also have their biological underpinnings in gut-mediated changes.
Overall, stress, anxiety and depression may be partly mediating their effects on hypertension and LVH, via increased gut permeability and decreased microbiome butyrate production. This increases systemic oxidative stress and pro-inflammatory cytokines that induce IDO and TDO, leading to the production of kynurenine and kynurenic acid, which activate the AhR and thereby increase CYP1B1 levels and activity that seem crucial to the pathophysiology of LVH in cardiomyocytes. The classical association of decreased serotonin in depression is strongly determined by such increases in IDO and TDO activity, including across a diverse range of medical conditions where depression is often comorbid [44], with the attenuated serotonin availability also driving down levels of melatonin and NAS. Incorporating CYP1B1 and the NAS/melatonin ratio into these gut-driven changes allows for a more plausible biological modelling of how stress and depression can link to hypertension and LVH (Figure 1).
6. Sirtuins, microRNAs, 14-3-3 and Mitochondria Functioning
CYP1B1 induction can lead to its translocation to mitochondria, where it mediates some of the toxicity associated with AhR ligands [45]. The importance of alterations in mitochondria functioning in LVH is highlighted by the data on the role of mitochondria-associated sirtuin-3 in LVH. In rodents, cardiac hypertrophy is attenuated by the inhibition of ROS and the induction of sirtuin-3 [46]. The maintenance of mitochondria sirtuin-3 attenuates both stress- and obesity-induced cardiac hypertrophy [47], highlighting the importance of sirtuin-3 and mitochondria functioning to the changes occurring in LVH. Cardiomyocyte cellular models also support a role for the loss of mitochondria sirtuin-3 in LVH [48].
Sirtuin-1 also plays a role in LVH, with sirtuin-1 alleles being risk factors for LVH in chronic kidney disease patients [49]. The efficacy of vitamin D3 against diabetes-associated cardiac hypertrophy is mediated by increased sirtuin-1, in association with attenuated DNA oxidative damage, decreased PARP1 and lower mammalian target of rapamycin (mTOR) phosphorylation [50]. As PARP1 is NAD+ dependent, it lowers the availability of the NAD+ that is necessary for sirtuin induction [51]. Protein kinase C (PKC)-ζ leads to cardiac hypertrophy via increased activity of nuclear factor-kappaB (NF-κB), ERK1/2 and ERK5, which sirtuin-1 prevents via PKC-ζ inhibition, as shown in preclinical models [52]. Sirtuin-1 is therefore a powerful regulator of many of the biological processes associated with LVH. As well as regulating mitochondria functioning via PGC-1α, which is decreased in LVH cardiomyocytes [53], sirtuin-1 can also positively regulate mitochondria via sirtuin-3. Sirtuin-1 deacetylases, and therefore positively regulates, mitochondria-associated sirtuin-3 [54]. Notably, sirtuin-3 is proposed to have co-evolved with melatonin within mitochondria, leading to their post-translational collaboration that, among other processes, acts to regulate mitochondria free radical generation and removal [55].
Sirtuin-3 also regulates mitochondria functioning and cardiac hypertrophy via the deacetylation of isocitrate dehydrogenase (IDH2). The IDH2 dimer links glucose metabolism to mitochondria oxidative phosphorylation [56]. A decrease in IDH2 activity leads to hypertrophy in cardiomyocyte cell lines in rodent LVH models [57][58], with the loss of IDH2 activity long appreciated to precede cardiac hypertrophy [59]. Sirtuin-3 is also a significant regulator of mitophagy, with dysregulated mitophagy being an important aspect of the biological underpinnings of LVH [60]. Such data highlight not only the importance of sirtuin regulation of IDH2, but also the crucial role that changes in mitochondria functioning play in the pathoetiology and pathophysiology of cardiac hypertrophy. There is a growing appreciation that mitochondria are an important hub for co-ordinating the receptor, ECM and intracellular signalling changes occurring in cells, with mitochondria driving the patterned responses and adaptations to such signalling, including in LVH [51][61].
A number of microRNAs show alterations in cardiac hypertrophy, including miR-7, miR-375 and miR-451 [62][63][64]. All three of these microRNAs can be increased in LVH and act to suppress 14-3-3 [65], with decreased 14-3-3 contributing to LVH pathophysiology [66]. It is of note that 14-3-3, miR-7, miR-375 and miR-451 can all act to regulate mitochondria functioning via alterations in the mitochondria melatonergic pathway [39]. This is mediated by 14-3-3 being necessary for the stabilization of arylalkylamine n-acetyltransferase (AANAT), which catalyses serotonin to NAS at the start of the melatonergic pathway. As such, the suppression of 14-3-3 by these miRNAs will significantly lower melatonergic pathway activity, which when coupled to increased CYP1B1 will dramatically lower melatonin availability (see Figure 1). However, it should be noted that not all studies show these three microRNAs to be increased in cardiac hypertrophy [67]. This may be a reflection on the wider dynamic and adaptive biological changes occurring in LVH, which rather than being a ‘static’ medical state may be better viewed as a series of plasticity adaptations.
7. Melatonin and LVH
A role for the melatonergic pathways in LVH is indicated by the data showing increased serotonin degradation by monoamine oxidase (MAO)A/B to be associated with LVH and wider cardiac dysregulation [68]. Serotonin is the necessary precursor of the melatonergic pathways, providing a substrate for AANAT to synthesize NAS, which is then enzymatically converted to melatonin by acetylserotonin methyltransferase (ASMT) [69]. Raised ROS and suboptimal mitochondria functioning are thought to increase MAO-A/B [70]. However, by decreasing the availability of serotonin as a substrate for the melatonergic pathways, the effects of raised MAO-A/B levels are likely to be, at least partly, mediated by the suppression of the mitochondria melatonergic pathways. Consequently, the lowered melatonin synthesis will contribute to an increase in ROS levels and suboptimal mitochondria functioning.
Given the plethora of studies showing the antioxidant, anti-inflammatory and mitochondria-optimizing effects of melatonin [43][55][61][71], a number of studies have investigated the utility of melatonin in LVH [72]. These authors showed melatonin to exert cardioprotective effects, reduce LVH remodelling and improve survival outcomes in the isoproterenol model of LVH [72]. Melatonin has clinical utility in the management of cardiac hypertrophy and affords protection in an array of different preclinical models, with effects variously attributed, including to an increase in PGC-1b [73], cyclophilin A/CD147 [74], antioxidant effects [75], retinoic acid receptor-related orphan receptor-α (RORα) [76], autophagy and AMPK [77] or a decrease in mTOR [78] and TNF-α [79]. Melatonin receptor regulating pharmaceuticals, such as ramelteon, have also shown utility in the management of LVH, suggesting a protective role for melatonin receptor activation [80].
However, it is important to note that a growing body of data shows melatonin to be produced by all mitochondria-containing cells so far investigated, usually within the mitochondria matrix [81], indicating that local melatonin synthesis and regulation may be an overlooked factor in many medical conditions [82], including glioblastoma [83], endometriosis [1] and neurodegenerative conditions [84]. The inclusion of the mitochondria melatonergic pathways in the pathophysiology of LVH allows the previously disparate bodies of data highlighted above to be integrated into a model of LVH that emphasizes the role of the mitochondria melatonergic pathways. The targeting of increased melatonin production in cardiomyocyte mitochondria may prove of far greater clinical utility than the administration of adjunctive melatonin.
References
- Anderson, G. Endometriosis Pathoetiology and Pathophsyiology: Roles of Vitamin A, Estrogen, Immunity, Adipocytes, Gut Microbiome and Melatonergic Pathway on Mitochondria Regulation. BioMol. Concepts 2019, 10, 133–149.
- Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 2164.
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol Rev. 2010, 90, 1507–1546.
- Zhang, B.; Zhang, P.; Tan, Y.; Feng, P.; Zhang, Z.; Liang, H.; Duan, W.; Jin, Z.; Wang, X.; Liu, J.; et al. C1q-TNF-related protein-3 attenuates pressure overload-induced cardiac hypertrophy by suppressing the p38/CREB pathway and p38-induced ER stress. Cell. Death. Dis. 2019, 10, 520.
- Martin, E.D.; Bassi, R.; Marber, M.S. p38 MAPK in cardioprotection - are we there yet? Br. J. Pharmacol. 2015, 172, 2101–2113.
- Marber, M.S.; Rose, B.; Wang, Y. The p38 mitogen-activated protein kinase pathway--a potential target for intervention in infarction, hypertrophy, and heart failure. J Mol Cell Cardiol. 2011, 51, 485–490.
- Li, K.L.; Lin, Y.C. PM2.5 induced cardiac hypertrophy via CREB/GSK3b/SOS1 pathway and metabolomics alterations. Oncotarget. 2018, 9, 30748–30760.
- Xiong, X.; Yang, X.; Duan, L.; Liu, W.; Zhang, Y.; Liu, Y.; Wang, P.; Li, S.; Li, X. Traditional Chinese medicine suppresses left ventricular hypertrophy by targeting extracellular signal-regulated kinases signaling pathway in spontaneously hypertensive rats. Sci. Rep. 2017, 7, 42965.
- Yang, H.; Feng, G.D.; Liang, Z.; Vitale, A.; Jiao, X.Y.; Ju, G.; You, S.W. In vitro beneficial activation of microglial cells by mechanically-injured astrocytes enhances the synthesis and secretion of BDNF through p38MAPK. Neurochem. Int. 2012, 61, 175–186.
- Nakaoka, Y.; Shioyama, W.; Kunimoto, S.; Arita, Y.; Higuchi, K.; Yamamoto, K.; Fujio, Y.; Nishida, K.; Kuroda, T.; Hirota, H.; et al. SHP2 mediates gp130-dependent cardiomyocyte hypertrophy via negative regulation of skeletal alpha-actin gene. J. Mol. Cell. Cardiol. 2010, 49, 157–164.
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Kunugi, H. Glucocorticoid suppresses BDNF-stimulated MAPK/ERK pathway via inhibiting interaction of Shp2 with TrkB. FEBS Lett. 2011, 585, 3224–3228.
- Jang, S.W.; Liu, X.; Pradoldej, S.; Tosini, G.; Chang, Q.; Iuvone, P.M.; Ye, K. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA 2010, 107, 3876–3881.
- Elkhatali, S.; Maayah, Z.H.; El-Sherbeni, A.A.; Elshenawy, O.H.; Abdelhamid, G.; Shoieb, S.M.; El-Kadi, A.O.S. Inhibition of Mid-chain HETEs Protects Against Angiotensin II-induced Cardiac Hypertrophy. J. Cardiovasc. Pharmacol. 2017, 70, 16–24.
- Alsaad, A.M.S. Dasatinib induces gene expression of CYP1A1, CYP1B1, and cardiac hypertrophy markers (BNP, β-MHC) in rat cardiomyocyte H9c2 cells. Toxicol. Mech. Methods 2018, 28, 678–684.
- Pingili, A.K.; Kara, M.; Khan, N.S.; Estes, A.M.; Lin, Z.; Li, W.; Gonzalez, F.J.; Malik, K.U. 6β-hydroxytestosterone, a cytochrome P450 1B1 metabolite of testosterone, contributes to angiotensin II-induced hypertension and its pathogenesis in male mice. Hypertension 2015, 65, 1279–1287.
- Jennings, B.L.; Montanez, D.E.; May, M.E., Jr.; Estes, A.M.; Fang, X.R.; Yaghini, F.A.; Kanu, A.; Malik, K.U. Cytochrome P450 1B1 contributes to increased blood pressure and cardiovascular and renal dysfunction in spontaneously hypertensive rats. Cardiovasc. Drugs. Ther. 2014, 28, 145–161.
- Zordoky, B.N.; El-Kadi, A.O. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and beta-naphthoflavone induce cellular hypertrophy in H9c2 cells by an aryl hydrocarbon receptor-dependant mechanism. Toxicol. In Vitro 2010, 24, 863–871.
- Zhou, B.; Wang, X.; Li, F.; Wang, Y.; Yang, L.; Zhen, X.; Tan, W. Mitochondrial activity and oxidative stress functions are influenced by the activation of AhR-induced CYP1A1 overexpression in cardiomyocytes. Mol. Med. Rep. 2017, 16, 174–180.
- Kopf, P.G.; Huwe, J.K.; Walker, M.K. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc. Toxicol. 2008, 8, 181–193.
- Yang, S.Y.; Ahmed, S.; Satheesh, S.V.; Matthews, J. Genome-wide mapping and analysis of aryl hydrocarbon receptor (AHR)- and aryl hydrocarbon receptor repressor (AHRR)-binding sites in human breast cancer cells. Arch. Toxicol. 2018, 92, 225–240.
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites With Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan Res. 2017, 10, 1178646917691738.
- Verheyen, N.; Meinitzer, A.; Grübler, M.R.; Ablasser, K.; Kolesnik, E.; Fahrleitner-Pammer, A.; Belyavskiy, E.; Trummer, C.; Schwetz, V.; Pieske-Kraigher, E.; et al. Low-grade inflammation and tryptophan-kynurenine pathway activation are associated with adverse cardiac remodeling in primary hyperparathyroidism: The EPATH trial. Clin. Chem. Lab. Med 2017, 55, 1034–1042.
- Mohan, M.; Al-Talabany, S.; McKinnie, A.; Mordi, I.R.; Singh, J.S.S.; Gandy, S.J.; Baig, F.; Hussain, M.S.; Bhalraam, U.; Khan, F.; et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: The MET-REMODEL trial. Eur. Heart. J. 2019, ehz203.
- Donelson, J.; Wang, Q.; Monroe, T.O.; Jiang, X.; Zhou, J.; Yu, H.; Mo, Q.; Sun, Q.; Marini, J.C.; Wang, X.; et al. Cardiac-specific ablation of glutaredoxin 3 leads to cardiac hypertrophy and heart failure. Physiol. Rep. 2019, 7, e14071.
- Martínez-Vieyra, V.; Rodríguez-Varela, M.; García-Rubio, D.; De la Mora-Mojica, B.; Méndez-Méndez, J.; Durán-Álvarez, C.; Cerecedo, D. Alterations to plasma membrane lipid contents affect the biophysical properties of erythrocytes from individuals with hypertension. Biochim. Biophys. Acta. Biomembr. 2019.
- Takahashi, K.; Murase, T.; Takatsu, M.; Matsuura, N.; Nagasawa, K.; Hattori, T.; Watanabe, S.; Murohara, T.; Nagata, K. Roles of oxidative stress and the mineralocorticoid receptor in cardiac pathology in a rat model of metabolic syndrome. Nagoya. J. Med. Sci. 2015, 77, 275–289.
- Krishnan, S.M.; Sobey, C.G.; Latz, E.; Mansell, A.; Drummond, G.R. IL-1β and IL-18: Inflammatory markers or mediators of hypertension? Br. J. Pharmacol. 2014, 171, 5589–5602.
- Nishikawa, K.; Yoshida, M.; Kusuhara, M.; Ishigami, N.; Isoda, K.; Miyazaki, K.; Ohsuzu, F. Left ventricular hypertrophy in mice with a cardiac-specific overexpression of interleukin-1. Am. J. Physiol. Heart. Circ. Physiol. 2006, 291, H176–H183.
- Morin, C.; Rousseau, E.; Blier, P.U.; Fortin, S. Effect of docosahexaenoic acid monoacylglyceride on systemic hypertension and cardiovascular dysfunction. Am. J. Physiol. Heart. Circ. Physiol. 2015, 309, H93–H102.
- Rudzite, V.K.; Vitols, A.V.; Liepinja, D.J.; Silava, A.K. Increased blood kynurenine level as a factor inhibiting the therapeutic effect of antihypertensive agents in combined long-term treatment of essential hypertension. Cor. Vasa. 1990, 32, 56–63.
- Anderson, G.; Ojala, J. Alzheimer’s and seizures: Interleukin-18, indoleamine 2,3-dioxygenase and quinolinic Acid. Int. J. Tryptophan Res. 2010, 3, 169–173.
- de Melo, L.G.P.; Nunes, S.O.V.; Anderson, G.; Vargas, H.O.; Barbosa, D.S.; Galecki, P.; Carvalho, A.F.; Maes, M. Shared metabolic and immune-inflammatory, oxidative and nitrosative stress pathways in the metabolic syndrome and mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 8, 34–50.
- Mocan, M.; Mocan Hognogi, L.D.; Anton, F.P.; Chiorescu, R.M.; Goidescu, C.M.; Stoia, M.A.; Farcas, A.D. Biomarkers of Inflammation in Left Ventricular Diastolic Dysfunction. Dis. Markers 2019, 2019, 7583690.
- Stanley, C.P.; Maghzal, G.J.; Ayer, A.; Talib, J.; Giltrap, A.M.; Shengule, S.; Wolhuter, K.; Wang, Y.; Chadha, P.; Suarna, C.; et al. Singlet molecular oxygen regulates vascular tone and blood pressure in inflammation. Nature 2019, 566, 548–552.
- Adams, S.; Teo, C.; McDonald, K.L.; Zinger, A.; Bustamante, S.; Lim, C.K.; Sundaram, G.; Braidy, N.; Brew, B.J.; Guillemin, G.J. Involvement of the kynurenine pathway in human glioma pathophysiology. PLoS ONE 2014, 9, e112945.
- Leshem, A.; Horesh, N.; Elinav, E. Fecal Microbial Transplantation and Its Potential Application in Cardiometabolic Syndrome. Front. Immunol. 2019, 10, 1341.
- Liu, M.Y.; Li, N.; Li, W.A.; Khan, H. Association between psychosocial stress and hypertension: A systematic review and meta-analysis. Neurol. Res. 2017, 39, 573–580.
- Tache, Y.; Larauche, M.; Yuan, P.Q.; Million, M. Brain and Gut CRF Signaling: Biological Actions and Role in the Gastrointestinal Tract. Curr. Mol. Pharmacol. 2018, 11, 51–71.
- Anderson, G. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Decreased Melatonin and Suboptimal Mitochondria Functioning: Pathoetiological and Pathophysiological Implications. Melatonin Res. 2019, 2, 70–85.
- Cruz Junho, C.V.; Trentin-Sonoda, M.; Alvim, J.M.; Gaisler-Silva, F.; Carneiro-Ramos, M.S. Ca2+/Calmodulin-dependent kinase II delta B is essential for cardiomyocyte hypertrophy and complement gene expression after LPS and HSP60 stimulation in vitro. Braz. J. Med. Biol. Res. 2019, 52, e8732.
- Martin-Subero, M.; Anderson, G.; Kanchanatawan, B.; Berk, M.; Maes, M. Comorbidity between depression and inflammatory bowel disease explained by immune-inflammatory, oxidative, and nitrosative stress; tryptophan catabolite; and gut-brain pathways. CNS Spectr. 2016, 21, 184–198.
- Takemura, Y.; Kikuchi, S.; Takagi, H.; Inaba, Y.; Nakagawa, K. A cross-sectional study on the relationship between depression and left ventricular hypertrophy. Prev. Med. 1998, 27, 787–791.
- Jin, C.J.; Engstler, A.J.; Sellmann, C.; Ziegenhardt, D.; Landmann, M.; Kanuri, G.; Lounis, H.; Schröder, M.; Vetter, W.; Bergheim, I. Sodium butyrate protects mice from the development of the early signs of non-alcoholic fatty liver disease: Role of melatonin and lipid peroxidation. Br. J. Nutr. 2016, 23, 1–12.
- Anderson, G.; Seo, M.; Berk, M.; Carvalho, A.F.; Maes, M. Gut Permeability and Microbiota in Parkinson’s Disease: Role of Depression, Tryptophan Catabolites, Oxidative and Nitrosative Stress and Melatonergic Pathways. Curr. Pharm. Des. 2016, 22, 6142–6151.
- Bansal, S.; Leu, A.N.; Gonzalez, F.J.; Guengerich, F.P.; Chowdhury, A.R.; Anandatheerthavarada, H.K.; Avadhani, N.G. Mitochondrial targeting of cytochrome P450 (CYP) 1B1 and its role in polycyclic aromatic hydrocarbon-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 9936–9951.
- Chen, Y.; Luo, H.Q.; Sun, L.L.; Xu, M.T.; Yu, J.; Liu, L.L.; Zhang, J.Y.; Wang, Y.Q.; Wang, H.X.; Bao, X.F.; et al. Dihydromyricetin Attenuates Myocardial Hypertrophy Induced by Transverse Aortic Constriction via Oxidative Stress Inhibition and SIRT3 Pathway Enhancement. Int. J. Mol. Sci. 2018, 19, 2592.
- Tronchere, H.; Cinato, M.; Timotin, A.; Guitou, L.; Villedieu, C.; Thibault, H.; Baetz, D.; Payrastre, B.; Valet, P.; Parini, A.; et al. Inhibition of PIKfyve prevents myocardial apoptosis and hypertrophy through activation of SIRT3 in obese mice. EMBO. Mol. Med. 2017, 9, 770–785.
- Lee, S.Y.; Ku, H.C.; Kuo, Y.H.; Yang, K.C.; Tu, P.C.; Chiu, H.L.; Su, M.J. Caffeic acid ethanolamide prevents cardiac dysfunction through sirtuin dependent cardiac bioenergetics preservation. J. Biomed. Sci. 2015, 22, 80.
- Spoto, B.; Ntounousi, E.; Testa, A.; Liakopoulos, V.; D’Arrigo, G.; Tripepi, G.; Parlongo, R.M.; Sanguedolce, M.C.; Mallamaci, F.; Zoccali, C. The sirtuin1 gene associates with left ventricular myocardial hypertrophy and remodeling in two chronic kidney disease cohorts: A longitudinal study. J. Hypertens. 2018, 36, 1705–1711.
- Qu, H.; Lin, K.; Wang, H.; Wei, H.; Ji, B.; Yang, Z.; Peng, C.; Xiao, X.; Deng, H. 1,25(OH)2 D3 improves cardiac dysfunction, hypertrophy, and fibrosis through PARP1/SIRT1/mTOR-related mechanisms in type 1 diabetes. Mol. Nutr. Food. Res. 2017, 61.
- Anderson, G. Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 255–266.
- Li, J.; Huang, J.; Lu, J.; Guo, Z.; Li, Z.; Gao, H.; Wang, P.; Luo, W.; Cai, S.; Hu, Y.; et al. Sirtuin 1 represses PKC-ζ activity through regulating interplay of acetylation and phosphorylation in cardiac hypertrophy. Br. J. Pharmacol. 2019, 176, 416–435.
- Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC- 1) regulatory cascade in cardiac physiology and disease. Circulation 2007, 115, 2540–2548.
- Kwon, S.; Seok, S.; Yau, P.; Li, X.; Kemper, B.; Kemper, J.K. Obesity and aging diminish sirtuin 1 (SIRT1)-mediated deacetylation of SIRT3, leading to hyperacetylation and decreased activity and stability of SIRT3. J. Biol. Chem. 2017, 292, 17312–17323.
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Jou, M.J.; Acuna-Castroviejo, D. Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3. Int. J. Mol. Sci. 2018, 9, 2439.
- Zou, X.; Zhu, Y.; Park, S.H.; Liu, G.; O’Brien, J.; Jiang, H.; Gius, D. SIRT3-Mediated Dimerization of IDH2 Directs Cancer Cell Metabolism and Tumor Growth. Cancer. Res. 2017, 77, 3990–3999.
- Ku, H.J.; Ahn, Y.; Lee, J.H.; Park, K.M.; Park, J.W. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free. Radic. Biol. Med. 2015, 80, 84–92.
- Lee., J.H.; Park, J.W. Attenuated mitochondrial NADP+-dependent isocitrate dehydrogenase activity induces apoptosis and hypertrophy of H9c2 cardiomyocytes. Biochimie 2014, 99, 110–118.
- Benderdour, M.; Charron, G.; DeBlois, D.; Comte, B.; Des Rosiers, C. Cardiac mitochondrial NADP+-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation: An event that precedes hypertrophy development. J. Biol. Chem. 2003, 278, 45154–45159.
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371.
- Polyakova, V.O.; Kvetnoy, I.M.; Anderson, G.; Rosati, J.; Mazzoccoli, G.; Linkova, N.S. Reciprocal Interactions of Mitochondria and the Neuroimmunoendocrine System in Neurodegenerative Disorders: An Important Role for Melatonin Regulation. Front. Physiol. 2018, 9, 199.
- Feng, H.; Wu, J.; Chen, P.; Wang, J.; Deng, Y.; Zhu, G.; Xian, J.; Huang, L.; Ouyang, W. MicroRNA-375-3p inhibitor suppresses angiotensin II-induced cardiomyocyte hypertrophy by promoting lactate dehydrogenase B expression. J. Cell. Physiol. 2019, 234, 14198–14209.
- Kaneto, C.M.; Nascimento, J.S.; Moreira, M.C.R.; Ludovico, N.D.; Santana, A.P.; Silva, R.A.A.; Silva-Jardim, I.; Santos, J.L.; Sousa, S.M.B.; Lima, P.S.P. MicroRNA profiling identifies miR-7-5p and miR-26b-5p as differentially expressed in hypertensive patients with left ventricular hypertrophy. Braz. J. Med. Biol. Res. 2017, 50, e6211.
- Kuwabara, Y.; Horie, T.; Baba, O.; Watanabe, S.; Nishiga, M.; Usami, S.; Izuhara, M.; Nakao, T.; Nishino, T.; Otsu, K.; et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ. Res. 2015, 116, 279–288.
- Anderson, G. Breast Cancer: Occluded Role of Mitochondria N-acetylserotonin/Melatonin Ratio in Co-ordinating Pathophysiology. Biochem. Pharmacol. 2019, 168, 259–268.
- Thandavarayan, R.A.; Giridharan, V.V.; Sari, F.R.; Arumugam, S.; Veeraveedu, P.T.; Pandian, G.N.; Palaniyandi, S.S.; Ma, M.; Suzuki, K.; Gurusamy, N.; et al. Depletion of 14-3-3 protein exacerbates cardiac oxidative stress, inflammation and remodeling process via modulation of MAPK/NF-ĸB signaling pathways after streptozotocin-induced diabetes mellitus. Cell. Physiol. Biochem. 2011, 28, 911–922.
- Song, L.; Su, M.; Wang, S.; Zou, Y.; Wang, X.; Wang, Y.; Cui, H.; Zhao, P.; Hui, R.; Wang, J. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 2014, 18, 2266–2274.
- Kaludercic, N.; Carpi, A.; Nagayama, T.; Sivakumaran, V.; Zhu, G.; Lai, E.W.; Bedja, D.; De Mario, A.; Chen, K.; Gabrielson, K.L.; et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid. Redox. Signal. 2014, 20, 267–280.
- Anderson, G.; Maes, M.; Berk, M. Schizophrenia is primed for an increased expression of depression through activation of immuno-inflammatory, oxidative and nitrosative stress, and tryptophan catabolite pathways. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 42, 101–114.
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; De Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: Role in chronic left ventricular dysfunction in mice. Antioxid. Redox. Signal. 2013, 18, 5–18.
- Reiter, R.J.; Manchester, L.C.; Fuentes-Broto, L.; Tan, D.X. Cardiac hypertrophy and remodelling: Pathophysiological consequences and protective effects of melatonin. J. Hypertens. 2010, 28 (Suppl 1), S7–S12.
- Simko, F.; Bednarova, K.R.; Krajcirovicova, K.; Hrenak, J.; Celec, P.; Kamodyova, N.; Gajdosechova, L.; Zorad, S.; Adamcova, M. Melatonin reduces cardiac remodeling and improves survival in rats with isoproterenol-induced heart failure. J. Pineal. Res. 2014, 57, 177–184.
- Zhai, M.; Liu, Z.; Zhang, B.; Jing, L.; Li, B.; Li, K.; Chen, X.; Zhang, M.; Yu, B.; Ren, K.; et al. Melatonin protects against the pathological cardiac hypertrophy induced by transverse aortic constriction through activating PGC-1β: In vivo and in vitro studies. J. Pineal. Res. 2017, 63.
- Su, H.; Li, J.; Chen, T.; Li, N.; Xiao, J.; Wang, S.; Guo, X.; Yang, Y.; Bu, P. Melatonin attenuates angiotensin II-induced cardiomyocyte hypertrophy through the CyPA/CD147 signaling pathway. Mol. Cell. Biochem. 2016, 422, 85–95.
- Ali, T.; Mushtaq, I.; Maryam, S.; Farhan, A.; Saba, K.; Jan, M.I.; Sultan, A.; Anees, M.; Duygu, B.; Hamera, S.; et al. Interplay of N acetyl cysteine and melatonin in regulating oxidative stress-induced cardiac hypertrophic factors and microRNAs. Arch. Biochem. Biophys. 2019, 661, 56–65.
- Xu, L.; Su, Y.; Zhao, Y.; Sheng, X.; Tong, R.; Ying, X.; Gao, L.; Ji, Q.; Gao, Y.; Yan, Y.; et al. Melatonin differentially regulates pathological and physiological cardiac hypertrophy: Crucial role of circadian nuclear receptor RORα signaling. J. Pineal. Res. 2019, 8, e12579.
- Xie, S.; Deng, Y.; Pan, Y.Y.; Wang, Z.H.; Ren, J.; Guo, X.L.; Yuan, X.; Shang, J.; Liu, H.G. Melatonin protects against chronic intermittent hypoxia-induced cardiac hypertrophy by modulating autophagy through the 5’ adenosine monophosphate-activated protein kinase pathway. Biochem. Biophys. Res. Commun. 2015, 464, 975–981.
- Kandemir, Y.B.; Tosun, V.; Güntekin, Ü. Melatonin protects against streptozotocin-induced diabetic cardiomyopathy through the mammalian target of rapamycin (mTOR) signaling pathway. Adv. Clin. Exp. Med. 2019.
- Lu, Q.; Yi, X.; Cheng, X.; Sun, X.; Yang, X. Melatonin protects against myocardial hypertrophy induced by lipopolysaccharide. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 353–360.
- Uchinaka, A.; Kawashima, Y.; Sano, Y.; Ito, S.; Sano, Y.; Nagasawa, K.; Matsuura, N.; Yoneda, M.; Yamada, Y.; Murohara, T.; et al. Effects of ramelteon on cardiac injury and adipose tissue pathology in rats with metabolic syndrome. Ann. N. Y. Acad. Sci. 2018, 1421, 73–87.
- Tan, D.X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal. Res. 2013, 54, 127–138.
- Anderson, G.; Maes, M. Local melatonin regulates inflammation resolution: A common factor in neurodegenerative, psychiatric and systemic inflammatory disorders. CNS. Neurol. Disord. Drug. Targets 2014, 13, 817–827.
- Anderson, G.; Reiter, R.J. Glioblastoma: Role of Mitochondria N-acetylserotonin/Melatonin Ratio in Mediating Effects of miR-451 and Aryl Hydrocarbon Receptor and in Coordinating Wider Biochemical Changes. Int. J. Tryptophan Res. 2019, 12.
- Anderson, G.; Maes, M. How Immune-inflammatory Processes Link CNS and Psychiatric Disorders: Classification and Treatment Implications. CNS. Neurol. Disord. Drug. Targets 2017, 16, 266–278.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
15 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No