 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Robert Eibl | + 2382 word(s) | 2382 | 2021-11-04 03:53:14 | | | |
| 2 | Conner Chen | Meta information modification | 2382 | 2021-11-15 03:06:11 | | | | |
| 3 | Robert Eibl | Meta information modification | 2382 | 2024-09-17 09:33:25 | | | | |
| 4 | Robert Eibl | Meta information modification | 2382 | 2024-09-17 09:39:59 | | | | |
| 5 | Robert Eibl | Meta information modification | 2382 | 2025-12-25 18:45:29 | | | | |
| 6 | Robert Eibl | Meta information modification | 2382 | 2025-12-25 18:55:30 | | | | |
| 7 | Robert Eibl | Meta information modification | 2382 | 2025-12-25 19:06:21 | | |
Video Upload Options
Direct biopsies obtain tissue material from the primary tumor, either via neurosurgical removal of all or most parts of a tumor or via stereotactic tissue biopsy. In contrast, a liquid biopsy uses body fluids collected distant to the brain tumor, such as venous blood from the arm or cerebrospinal fluid (CSF) via lumbar or cisternal puncture.
[1]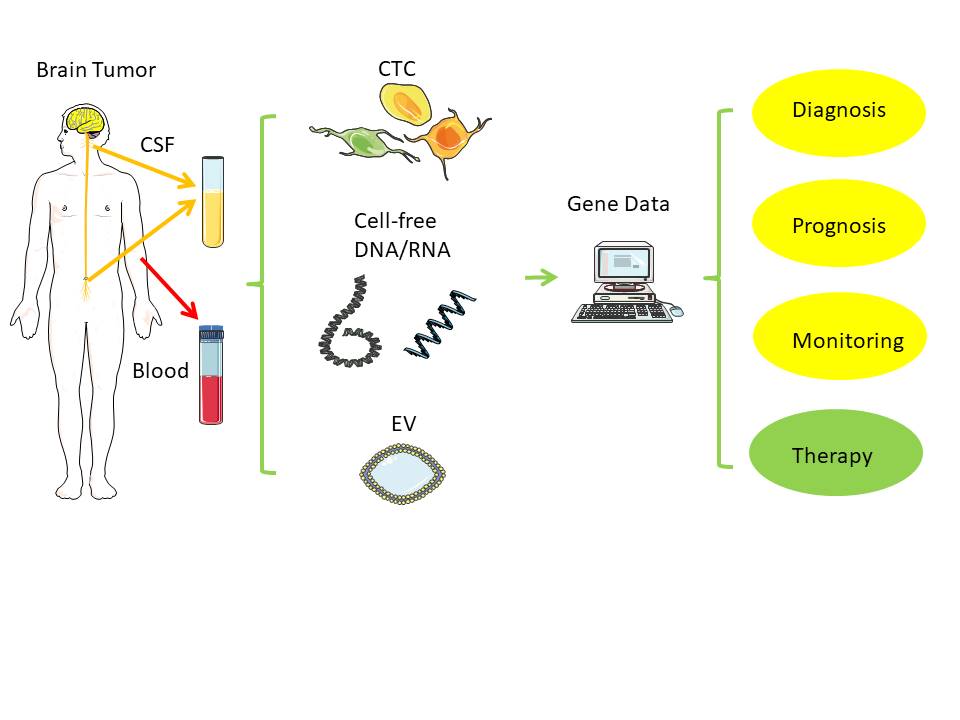
Figure 1. Liquid biopsy. Distant from the original brain tumor, samples from blood and cerebrospinal fluid (CSF) can typically serve as low-risk source of tumor-derived nucleic acids (RNA, DNA) for further analysis[1]. Notes: CSF - cerebrospinal fluid; EV - extracellular vesicle; CTC - circulating tumor cell. Created/modified with https://smart.servier.com (accessed on 8 August 2021), licensed under Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/ (accessed on 8 August 2021)).
1. Circulating Tumor Cells—CTCs
2. Cell-Free DNA and Circulating Tumor DNA
| Year | Gene | Variation | Source | Method | Tumor |
|---|---|---|---|---|---|
| 2003 [38] | MGMT (promoter) |
Methylation | Serum | MS-PCR | GBM |
| 2006 [44] | Methylation | Plasma | MS-PCR | GBM, AA | |
| 2010 [40] | Methylation | Serum | MS-PCR | Astrocytic tumors (WHO III, IV), oligodendroglial tumors (WHO II, III) |
|
| 2013 [41] | Methylation | Serum | MS-PCR | Glial tumors (II, III, IV), meningioma | |
| 2003 [38] | p16 | Methylation | Serum | MS-PCR | GBM |
| 2006 [44] | Methylation | Plasma | MS-PCR | GBM, AA, AOA | |
| 2003 [38] | DAPK | Methylation | Serum | MS-PCR | GBM |
| 2003 [38] | RASSF1A | Methylation | Serum | MS-PCR | GBM |
| 2013 [41] | Methylation | Serum | MS-PCR | Glial tumors (II, III, IV), meningioma | |
| 2006 [44] | p73 | Methylation | Plasma | MS-PCR | GBM |
| 2010 [40] | PTEN | Methylation | MS-PCR | MS-PCR | Astrocytic tumors (WHO III, IV) |
| 2014 [39] | Mutation | Plasma, serum | Digital PCR, sequencing |
Glioma II, AA, GBM | |
| 2010 [40] | 10q | LOH | Serum | LOH | Astrocytic (WHO III, IV), Oligodendroglial (WHO II, III) |
| 2010 [40] | 1p | LOH | Serum | LOH | Oligodendroglial (WHO II, III) |
| 2010 [40] | 19q | LOH | Serum | LOH | Oligodendroglial (WHO II, III) |
| 2012 [42] | IDH1 | Mutation (R132H) | Plasma | digital PCR | Glioma (WHO grade II, III, IV) |
| 2014 [39] | Mutation | Plasma, serum | Digital PCR, sequencing |
Glioma II, AA, GBM | |
| 2013 [41] | p15INK4B | Methylation | Serum | MS-PCR | Glial tumors (II, III, IV), meningioma |
| 2013 [41] | p14ARF | Methylation | Serum | MS-PCR | Glial tumors (II, III, IV), meningioma |
| 2014 [39] | TP53 | Mutation | Plasma, serum | Digital PCR, sequencing |
Glioma II, AA, GBM |
| 2014 [39] | EGFR | Mutations | Plasma, serum | Digital PCR, sequencing |
Glioma II, AA, GBM |
| 2014 [39] | PIK3CA | Mutation | Plasma, serum | Digital PCR, sequencing |
Glioma II, AA, GBM |
| 2015 [53] | TP53 (R114C) EPHB1 TERT PIK3CG IDH1 (R132H) ANK (K2337X) EGFR (C620S) PTEN (D162) FTH1 (R108K) OR51D1 (R135C) |
Mutations | CSF, (plasma) | ddPCR, MAF | GBM |
| 2015 [52] | Genome | Mutations | CSF | TAS/WES | AA III, PA I, ependymoma, medulloblastoma IV, GBM, LGG II, diffuse astrocytoma |
| 2015 [51] | Gene panel (587 genes) including NF2, AKT1 |
Mutations | CSF, plasma, serum | ddPCR/TAS | Vestibular schwannoma, atypical meningioma |
| 2017 [43] | Gene panels (54, 68, 70 genes) including p53, EGFR, Met |
Mutations | Plasma | NGS | Brain tumors (not specified) |
| 2018 [48] | IDH1, IDH2, TP53, TERT, ATRX, H3F3A, HIST1H3B | Mutations | CSF | sequencing | Diffuse gliomas |
| 2018 [50] | Genome | SCNAs and fragmentation |
CSF | WGS | Glioma |
| 2018 [62] | TERT | Mutation | CSF, (plasma) | PCR, sequencing | GBM |
| 2019 [63] | BRAF | Mutation (V600E) | CSF, plasma, serum | dPCR | PXA, ganglioglioma, PA, pilomyxoid astrocytoma |
| 2019 [46] | Genome including TP53, JAK2, NF1, EGFR, BRAF, IDH1, NRAS, GNAS, ATM | Mutations | Plasma | NGS | Astrocytic/oligodendral tumors grades I–IV, including GBM, medulloblastoma, meningioma, and ependymoma |
| 2019 [49] | IDH1 1P19Q CIC ATRX TP53 |
Mutations | CSF | NGS | LGG, GBM |
3. MicroRNA—miRNA—miR
| Year | miR | Variation | Source | Method | Tumor |
|---|---|---|---|---|---|
| 2016 [85] | miR-10-b | Up/progression | Serum | qPCR | HGG |
| miR-21 | |||||
| 2016 [74] | miR-205 | Down/diagnostics | Serum | qPCR | Glioma |
| 2018 [71] | Panel of 7 miRNAs | Diagnostic signature | Serum EV | NGS | GBM |
| 2020 [69] | miR-21 | Up/progression | Serum | ddPCR | LGG, GBM |
| miR-20e | |||||
| miR-223 | |||||
| 2020 [70] | miR-17-5p | Up/progression | Serum | qPCR | GBM |
| miR-125b | |||||
| miR-221 | |||||
| 2020 [88] | miR-486 | Up/diagnostic | EV from tumor microenvironment/neurosurgical aspirate fluid | NGS | GBM |
| 2021 [89] | miR-21 | Up/progression | Serum EV | qPCR | HGG |
| miR124-3p | |||||
| miR-222 | |||||
| 2021 [87] | Panel of 23 miRNAs | Screening signature | Urine | nanowire | GBM, glioma |
4. Extracellular Vesicles—EVs
References
- Robert H. Eibl; Markus Schneemann. Liquid Biopsy and Cancer; Springer Nature: Cham, Switzerland, 2024; pp. 1-25.
- Eibl, R.H., Schneemann, M. Liquid Biopsy and Cancer. In: Cancer Treatment Modalities: An Interdisciplinary Approach.; Rezaei, N., Eds.; Springer Nature: Cham, Switzerland, 2025; pp. 367-391.
- Eibl, R.H.; P. Kleihues; P. S. Jat; O. D. Wiestler; A model for primitive neuroectodermal tumors in transgenic neural transplants harboring the SV40 large T antigen.. The American Journal of Pathology 1994, 144, 556-564.
- Wiestler, O.D.; Aguzzi, A.; Schneemann, M.; Eibl, R.; von Deimling, A.; Kleihues, P; Oncogene complementation in fetal brain transplants.. Cancer Research 1992, 52, 3760-3767.
- Radner, H.; El-Shabrawi, Y.; Eibl, R.H.; Brüstle, O.; Kenner, L.; Kleihues, P; Wiestler, O.D.; Tumor induction by ras and myc oncogenes in fetal and neonatal brain: modulating effects of developmental stage and retroviral dose. Acta Neuropathologica 1993, 86, 456-465.
- Wiestler, O.D.; Brüstle, O.; Eibl, R.H.; Radner, H.; von Deimling, A.; Plate, K.; Aguzzi, A.; Kleihues, P.; A new approach to the molecular basis of neoplastic transformation in the brain. Neuropathology and Applied Neurobiology 1992, 18, 443-453.
- Wiestler, O.D.; Brüstle, O.; Eibl, R.H.; Radner, H.; Aguzzi, A.; Kleihues, P.; Retrovirus-mediated oncogene transfer into neural transplants.. Brain Pathology 1992, 2, 47-59.
- Wiestler, O.D.; Brüstle, O.; Eibl, R.H.; Radner, H.; Aguzzi, A.; Kleihues, P.; Oncogene Transfer into the Brain. Recent Results Cancer Res. 1994, 135, 55-66.
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain Tumor Cells in Circulation Are Enriched for Mesenchymal Gene Expression. Cancer Discov. 2014, 4, 1299–1309.
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous Dissemination of Glioblastoma Multiforme. Sci. Transl. Med. 2014, 6, 247ra101.
- Perryman, L.; Erler, J.T. Brain Cancer Spreads. Sci. Transl. Med. 2014, 6, 247fs28.
- Macarthur, K.M.; Kao, G.D.; Chandrasekaran, S.; Alonso-Basanta, M.; Chapman, C.; Lustig, R.A.; Wileyto, E.P.; Hahn, S.M.; Dorsey, J.F. Detection of Brain Tumor Cells in the Peripheral Blood by a Telomerase Promoter-Based Assay. Cancer Res. 2014, 74, 2152–2159.
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.M.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of Circulating Tumour Cell Clusters in Human Glioblastoma. Br. J. Cancer 2018, 119, 487–491.
- Gao, F.; Cui, Y.; Jiang, H.; Sui, D.; Wang, Y.; Jiang, Z.; Zhao, J.; Lin, S. Circulating Tumor Cell Is a Common Property of Brain Glioma and Promotes the Monitoring System. Oncotarget 2016, 7, 71330–71340.
- Robert H. Eibl; Markus Schneemann; Circulating Tumor Cells in Glioblastoma. Cancers. 2025, 18, 10.
- Eibl, R.H.; Pietsch, T.; Moll, J.; Skroch-Angel, J; Heider, K.H.; von Ammon, K.; Wiestler, O.D.; Ponta, H.; Kleihues, P.; Herrlich, P.; et al. Expression of variant CD44 epitopes in human astrocytic brain tumors. Journal of Neuro-Oncology 1995, 26, 165-170.
- Paget, S. The Distribution of Secondary Growths in Cancer of the Breast. Lancet 1889, 133, 571–573.
- Gonzalez-Beltran, A.N.; Masuzzo, P.; Ampe, C.; Bakker, G.-J.; Besson, S.; Eibl, R.H.; Friedl, P.; Gunzer, M.; Kittisopikul, M.; Dévédec, S.E.L.; et al. Community standards for open cell migration data. GigaScience 2020, 9, giaa041.
- Eibl, R.H.; Benoit, M; Molecular resolution of cell adhesion forces. IEE Proceedings - Nanobiotechnology 2004, 151, 128-132.
- Eibl, R.H.; Moy, V.T.; Atomic Force Microscopy Measurements of Protein–Ligand Interactions on Living Cells. Protein-Ligand Interactions 2005, 305, 439-450.
- Eibl, R.H.; Comment on “A method to measure cellular adhesion utilizing a polymer micro-cantilever” [Appl. Phys. Lett. 103, 123702 (2013)]. Applied Physics Letters 2014, 104, 236103.
- Eibl, R.H.; Single-Molecule Studies of Integrins by AFM-Based Force Spectroscopy on Living Cells. Scanning Probe Microscopy in Nanoscience and Nanotechnology 3 2012, 3, 137-169.
- Pax, M.; Rieger, J.; Eibl, R.H.; Thielemann, C.; Johannsmann, D.; Measurements of fast fluctuations of viscoelastic properties with the quartz crystal microbalance. The Analyst 2005, 130, 1474-1477.
- Eibl, R.H.; Cell Adhesion Receptors Studied by AFM-Based Single-Molecule Force Spectroscopy. Scanning Probe Microscopy in Nanoscience and Nanotechnology 2 2010, 2, 197-215.
- Eibl, R.H.; Moy, V.T.; AFM-based adhesion measurement of single receptor-ligand bonds on living cells. In: Recent Res. Devel. Biophys., 3(2004), 235-246, ISBN: 81-7895-130-4, Transworld Research Network, Kerala
- Eibl, R.H.; Direct Force Measurements of Receptor-Ligand Interactions on living cells. In: Applied Scanning Probe Methods XII - Characterization. Bhushan B, Fuchs H (Editors), Springer, pp. 1-31, (2009)
- Eibl R.H.; First measurement of physiologic VLA-4 activation by SDF-1 at the single-molecule level on a living cell. In: Advances in Single Molecule Research for Biology and Nanoscience. Hinterdorfer P, Schuetz G, Pohl P (Editors),Trauner, Linz, p40-43, ISBN 3854991630 (2007)
- Eibl, R.H.; Atomic force microscopy measurement of SDF-1 mediated affinity modulation of single VLA-4 - VCAM-1 bonds. In: Immunology 2004: Cytokine Network, Regulatory Cells, Signaling and Apoptosis. Monduzzi Ed. (Editor: E. Skamene), MEDIMOND S.r.l., Bologna, Italy, (1):115-120 ISBN 978-88-7587-070-6 (2004)
- Pantel, K.; Denève, E.; Nocca, D.; Coffy, A.; Vendrell, J.-P.; Maudelonde, T.; Riethdorf, S.; Alix-Panabières, C. Circulating Epithelial Cells in Patients with Benign Colon Diseases. Clin. Chem. 2012, 58, 936–940.
- Mazard, T.; Cayrefourcq, L.; Perriard, F.; Senellart, H.; Linot, B.; de la Fouchardière, C.; Terrebonne, E.; François, E.; Obled, S.; Guimbaud, R.; et al. Clinical Relevance of Viable Circulating Tumor Cells in Patients with Metastatic Colorectal Cancer: The COLOSPOT Prospective Study. Cancers 2021, 13, 2966.
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243.
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating Mutant DNA to Assess Tumor Dynamics. Nat. Med. 2008, 14, 985–990.
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162.
- Ohgaki, H; Eibl, R.H.; Wiestler, O.D.; Yasargil, M.G.; Newcom, E.W.; Kleihues, P.; p53 mutations in nonastrocytic human brain tumors.. Cancer Research 1991, 51, 6202-6205.
- von Deimling, A.; Eibl, R.H.; Ohgaki, H.; Louis, D.N.; von Ammon, K.; Petersen, I.; Kleihues, P.; Chung, R.Y.; Wiestler, O.D.; Seizinger, B.R.; et al. P53 Mutations Are Associated with 17p Allelic Loss in Grade II and Grade III Astrocytoma.. Cancer Res. 1992, 52, 2987-2990.
- Ohgaki, H.; Eibl, R.H.; Schwab, M.; Reichel, M.B.; Mariani, L.; Gehring, M.; Petersen, I.; Höll, T.; Wiestler, O.D.; Kleihues, P.; et al. Mutations of thep53 tumor suppressor gene in neoplasms of the human nervous system. Molecular Carcinogenesis 1993, 8, 74-80.
- Kleihues, P.; Ohgaki, H.; Eibl, R.H.; Reichel, M.B.; Mariani, L.; Gehring, M.; Petersen, I.; Höll, T.; von Deimling, A.; Wiestler, O.D.; et al. Type and Frequency of p53 Mutations in Tumors of the Nervous System and Its Coverings. Methods in Molecular Biology 1994, 135, 25-31.
- Balaña, C.; Ramirez, J.L.; Taron, M.; Roussos, Y.; Ariza, A.; Ballester, R.; Sarries, C.; Mendez, P.; Sanchez, J.J.; Rosell, R. O6-Methyl-Guanine-DNA Methyltransferase Methylation in Serum and Tumor DNA Predicts Response to 1,3-Bis(2-Chloroethyl)-1-Nitrosourea but Not to Temozolamide plus Cisplatin in Glioblastoma Multiforme. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1461–1468.
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24.
- Lavon, I.; Refael, M.; Zelikovitch, B.; Shalom, E.; Siegal, T. Serum DNA Can Define Tumor-Specific Genetic and Epigenetic Markers in Gliomas of Various Grades. Neuro-Oncology 2010, 12, 173–180.
- Majchrzak-Celińska, A.; Paluszczak, J.; Kleszcz, R.; Magiera, M.; Barciszewska, A.-M.; Nowak, S.; Baer-Dubowska, W. Detection of MGMT, RASSF1A, P15INK4B, and P14ARF Promoter Methylation in Circulating Tumor-Derived DNA of Central Nervous System Cancer Patients. J. Appl. Genet. 2013, 54, 335–344.
- Boisselier, B.; Gallego Perez-Larraya, J.; Rossetto, M.; Labussiere, M.; Ciccarino, P.; Marie, Y.; Delattre, J.-Y.; Sanson, M. Detection of IDH1 Mutation in the Plasma of Patients with Glioma. Neurology 2012, 79, 1693–1698.
- Schwaederle, M.; Chattopadhyay, R.; Kato, S.; Fanta, P.T.; Banks, K.C.; Choi, I.S.; Piccioni, D.E.; Ikeda, S.; Talasaz, A.; Lanman, R.B.; et al. Genomic Alterations in Circulating Tumor DNA from Diverse Cancer Patients Identified by Next-Generation Sequencing. Cancer Res. 2017, 77, 5419–5427.
- Weaver, K.D.; Grossman, S.A.; Herman, J.G. Methylated Tumor-Specific DNA as a Plasma Biomarker in Patients with Glioma. Cancer Investig. 2006, 24, 35–40.
- Bagley, S.J.; Nabavizadeh, S.A.; Mays, J.J.; Till, J.E.; Ware, J.B.; Levy, S.; Sarchiapone, W.; Hussain, J.; Prior, T.; Guiry, S.; et al. Clinical Utility of Plasma Cell-Free DNA in Adult Patients with Newly Diagnosed Glioblastoma: A Pilot Prospective Study. Clin. Cancer Res. 2020, 26, 397–407.
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of Cell-Free Circulating Tumor DNA in 419 Patients with Glioblastoma and Other Primary Brain Tumors. CNS Oncol. 2019, 8, CNS34.
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538.
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M.A.; López-Bigas, N.; y Cajal, S.R.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin. Cancer Res. 2018, 24, 2812–2819.
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking Tumour Evolution in Glioma through Liquid Biopsies of Cerebrospinal Fluid. Nature 2019, 565, 654–658.
- Mouliere, F.; Mair, R.; Chandrananda, D.; Marass, F.; Smith, C.G.; Su, J.; Morris, J.; Watts, C.; Brindle, K.M.; Rosenfeld, N. Detection of Cell-free DNA Fragmentation and Copy Number Alterations in Cerebrospinal Fluid from Glioma Patients. EMBO Mol. Med. 2018, 10, e9323.
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain Tumor Mutations Detected in Cerebral Spinal Fluid. Clin. Chem. 2015, 61, 514–522.
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of Tumor-Derived DNA in Cerebrospinal Fluid of Patients with Primary Tumors of the Brain and Spinal Cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709.
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal Fluid-Derived Circulating Tumour DNA Better Represents the Genomic Alterations of Brain Tumours than Plasma. Nat. Commun. 2015, 6, 8839.
- Huang, T.Y.; Piunti, A.; Lulla, R.R.; Qi, J.; Horbinski, C.M.; Tomita, T.; James, C.D.; Shilatifard, A.; Saratsis, A.M. Detection of Histone H3 Mutations in Cerebrospinal Fluid-Derived Tumor DNA from Children with Diffuse Midline Glioma. Acta Neuropathol. Commun. 2017, 5, 28.
- Robert H. Eibl; Markus Schneemann; Medulloblastoma: From TP53 Mutations to Molecular Classification and Liquid Biopsy. Biol.. 2023, 12, 267.
- Robert H. Eibl; Markus Schneemann; Liquid biopsy for monitoring medulloblastoma. Extracell. Vesicles Circ. Nucleic Acids. 2022, 3, 263-74.
- Floyd, D.; Purow, B. Micro-Masters of Glioblastoma Biology and Therapy: Increasingly Recognized Roles for MicroRNAs. Neuro-Oncology 2014, 16, 622–627.
- Ding, Y.; Lu, C.; Zhang, W.; Wang, Y.; Li, Y.; Zhu, Y.; Lv, S.; Zhang, J. The Emerging Role of Circular RNAs in Cardiovascular Diseases. J. Physiol. Biochem. 2021, 77, 343–353.
- Chen, W.W.; Balaj, L.; Liau, L.M.; Samuels, M.L.; Kotsopoulos, S.K.; Maguire, C.A.; Loguidice, L.; Soto, H.; Garrett, M.; Zhu, L.D.; et al. BEAMing and Droplet Digital PCR Analysis of Mutant IDH1 MRNA in Glioma Patient Serum and Cerebrospinal Fluid Extracellular Vesicles. Mol. Ther. Nucleic Acids 2013, 2, e109.
- Preuss, I.; Eberhagen, I.; Haas, S.; Eibl, R.H.; Kaufmann, M.; von Minckwitz, G.; Kaina, B. O6-Methylguanine-DNA Methyltransferase Activity in Breast and Brain Tumors. Int. J. Cancer 1995, 61, 321–326.
- Preuss, I.; Haas, S.; Eichhorn, U.; Eberhagen, I.; Kaufmann, M.; Beck, T.; Eibl, R.H.; Dall, P.; Bauknecht, T.; Hengstler, J.; et al. Activity of the DNA Repair Protein O6-Methylguanine-DNA Methyltransferase in Human Tumor and Corresponding Normal Tissue. Cancer Detect. Prev. 1996, 20, 130–136.
- Juratli, T.A.; Stasik, S.; Zolal, A.; Schuster, C.; Richter, S.; Daubner, D.; Juratli, M.A.; Thowe, R.; Hennig, S.; Makina, M.; et al. TERT Promoter Mutation Detection in Cell-Free Tumor-Derived DNA in Patients with IDH Wild-Type Glioblastomas: A Pilot Prospective Study. Clin. Cancer Res. 2018, 24, 5282–5291.
- García-Romero, N.; Carrión-Navarro, J.; Areal-Hidalgo, P.; Ortiz de Mendivil, A.; Asensi-Puig, A.; Madurga, R.; Núñez-Torres, R.; González-Neira, A.; Belda-Iniesta, C.; González-Rumayor, V.; et al. BRAF V600E Detection in Liquid Biopsies from Pediatric Central Nervous System Tumors. Cancers 2019, 12, 66.
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854.
- Jones, J.; Nguyen, H.; Drummond, K.; Morokoff, A. Circulating Biomarkers for Glioma: A Review. Neurosurgery 2021, 88, E221–E230.
- Roth, P.; Wischhusen, J.; Happold, C.; Chandran, P.A.; Hofer, S.; Eisele, G.; Weller, M.; Keller, A. A Specific MiRNA Signature in the Peripheral Blood of Glioblastoma Patients. J. Neurochem. 2011, 118, 449–457.
- Wang, Q.; Li, P.; Li, A.; Jiang, W.; Wang, H.; Wang, J.; Xie, K. Plasma Specific MiRNAs as Predictive Biomarkers for Diagnosis and Prognosis of Glioma. J. Exp. Clin. Cancer Res. 2012, 31, 97.
- Yang, C.; Wang, C.; Chen, X.; Chen, S.; Zhang, Y.; Zhi, F.; Wang, J.; Li, L.; Zhou, X.; Li, N.; et al. Identification of Seven Serum MicroRNAs from a Genome-Wide Serum MicroRNA Expression Profile as Potential Noninvasive Biomarkers for Malignant Astrocytomas. Int. J. Cancer 2013, 132, 116–127.
- Morokoff, A.; Jones, J.; Nguyen, H.; Ma, C.; Lasocki, A.; Gaillard, F.; Bennett, I.; Luwor, R.; Stylli, S.; Paradiso, L.; et al. Serum MicroRNA Is a Biomarker for Post-Operative Monitoring in Glioma. J. Neurooncol. 2020, 149, 391–400.
- Swellam, M.; Bakr, N.M.; El Magdoub, H.M.; Hamza, M.S.; Ezz El Arab, L.R. Emerging Role of MiRNAs as Liquid Biopsy Markers for Prediction of Glioblastoma Multiforme Prognosis. J. Mol. Neurosci. 2021, 71, 836–844.
- Ebrahimkhani, S.; Vafaee, F.; Hallal, S.; Wei, H.; Lee, M.Y.T.; Young, P.E.; Satgunaseelan, L.; Beadnall, H.; Barnett, M.H.; Shivalingam, B.; et al. Deep Sequencing of Circulating Exosomal MicroRNA Allows Non-Invasive Glioblastoma Diagnosis. NPJ Precis. Oncol. 2018, 2, 28.
- Zhao, H.; Shen, J.; Hodges, T.R.; Song, R.; Fuller, G.N.; Heimberger, A.B. Serum MicroRNA Profiling in Patients with Glioblastoma: A Survival Analysis. Mol. Cancer 2017, 16, 59.
- Huang, Q.; Wang, C.; Hou, Z.; Wang, G.; Lv, J.; Wang, H.; Yang, J.; Zhang, Z.; Zhang, H. Serum MicroRNA-376 Family as Diagnostic and Prognostic Markers in Human Gliomas. Cancer Biomark. Sect. Dis. Markers 2017, 19, 137–144.
- Yue, X.; Lan, F.; Hu, M.; Pan, Q.; Wang, Q.; Wang, J. Downregulation of Serum MicroRNA-205 as a Potential Diagnostic and Prognostic Biomarker for Human Glioma. J. Neurosurg. 2016, 124, 122–128.
- Xiao, Y.; Zhang, L.; Song, Z.; Guo, C.; Zhu, J.; Li, Z.; Zhu, S. Potential Diagnostic and Prognostic Value of Plasma Circulating MicroRNA-182 in Human Glioma. Med. Sci. Monit. 2016, 22, 855–862.
- Lai, N.-S.; Wu, D.-G.; Fang, X.-G.; Lin, Y.-C.; Chen, S.-S.; Li, Z.-B.; Xu, S.-S. Serum MicroRNA-210 as a Potential Noninvasive Biomarker for the Diagnosis and Prognosis of Glioma. Br. J. Cancer 2015, 112, 1241–1246.
- Zhi, F.; Shao, N.; Wang, R.; Deng, D.; Xue, L.; Wang, Q.; Zhang, Y.; Shi, Y.; Xia, X.; Wang, S.; et al. Identification of 9 Serum MicroRNAs as Potential Noninvasive Biomarkers of Human Astrocytoma. Neuro-Oncology 2015, 17, 383–391.
- Sun, J.; Liao, K.; Wu, X.; Huang, J.; Zhang, S.; Lu, X. Serum MicroRNA-128 as a Biomarker for Diagnosis of Glioma. Int. J. Clin. Exp. Med. 2015, 8, 456–463.
- Manterola, L.; Guruceaga, E.; Gállego Pérez-Larraya, J.; González-Huarriz, M.; Jauregui, P.; Tejada, S.; Diez-Valle, R.; Segura, V.; Samprón, N.; Barrena, C.; et al. A Small Noncoding RNA Signature Found in Exosomes of GBM Patient Serum as a Diagnostic Tool. Neuro-Oncol. 2014, 16, 520–527.
- Wu, J.; Li, L.; Jiang, C. Identification and Evaluation of Serum MicroRNA-29 Family for Glioma Screening. Mol. Neurobiol. 2015, 52, 1540–1546.
- Ivo D’Urso, P.; Fernando D’Urso, O.; Damiano Gianfreda, C.; Mezzolla, V.; Storelli, C.; Marsigliante, S. MiR-15b and MiR-21 as Circulating Biomarkers for Diagnosis of Glioma. Curr. Genom. 2015, 16, 304–311.
- Zhang, R.; Pang, B.; Xin, T.; Guo, H.; Xing, Y.; Xu, S.; Feng, B.; Liu, B.; Pang, Q. Plasma MiR-221/222 Family as Novel Descriptive and Prognostic Biomarkers for Glioma. Mol. Neurobiol. 2016, 53, 1452–1460.
- Wei, X.; Chen, D.; Lv, T.; Li, G.; Qu, S. Serum MicroRNA-125b as a Potential Biomarker for Glioma Diagnosis. Mol. Neurobiol. 2016, 53, 163–170.
- Shao, N.; Wang, L.; Xue, L.; Wang, R.; Lan, Q. Plasma MiR-454-3p as a Potential Prognostic Indicator in Human Glioma. Neurol. Sci. 2014, 36, 309–313.
- Siegal, T.; Charbit, H.; Paldor, I.; Zelikovitch, B.; Canello, T.; Benis, A.; Wong, M.L.; Morokoff, A.P.; Kaye, A.H.; Lavon, I. Dynamics of Circulating Hypoxia-Mediated MiRNAs and Tumor Response in Patients with High-Grade Glioma Treated with Bevacizumab. J. Neurosurg. 2016, 125, 1008–1015.
- Ali, H.; Harting, R.; de Vries, R.; Ali, M.; Wurdinger, T.; Best, M.G. Blood-Based Biomarkers for Glioma in the Context of Gliomagenesis: A Systematic Review. Front. Oncol. 2021, 11, 2134.
- Kitano, Y.; Aoki, K.; Ohka, F.; Yamazaki, S.; Motomura, K.; Tanahashi, K.; Hirano, M.; Naganawa, T.; Iida, M.; Shiraki, Y.; et al. Urinary MicroRNA-Based Diagnostic Model for Central Nervous System Tumors Using Nanowire Scaffolds. ACS Appl. Mater. Interfaces 2021, 13, 17316–17329.
- Hallal, S.; Ebrahim Khani, S.; Wei, H.; Lee, M.Y.T.; Sim, H.-W.; Sy, J.; Shivalingam, B.; Buckland, M.E.; Alexander-Kaufman, K.L. Deep Sequencing of Small RNAs from Neurosurgical Extracellular Vesicles Substantiates MiR-486-3p as a Circulating Biomarker That Distinguishes Glioblastoma from Lower-Grade Astrocytoma Patients. Int. J. Mol. Sci. 2020, 21, 4954.
- Olioso, D.; Caccese, M.; Santangelo, A.; Lippi, G.; Zagonel, V.; Cabrini, G.; Lombardi, G.; Dechecchi, M.C. Serum Exosomal MicroRNA-21, 222 and 124-3p as Noninvasive Predictive Biomarkers in Newly Diagnosed High-Grade Gliomas: A Prospective Study. Cancers 2021, 13, 3006.
- Figueroa, J.M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of Wild-Type EGFR Amplification and EGFRvIII Mutation in CSF-Derived Extracellular Vesicles of Glioblastoma Patients. Neuro-Oncology 2017, 19, 1494–1502.
- Manda, S.V.; Kataria, Y.; Tatireddy, B.R.; Ramakrishnan, B.; Ratnam, B.G.; Lath, R.; Ranjan, A.; Ray, A. Exosomes as a Biomarker Platform for Detecting Epidermal Growth Factor Receptor–Positive High-Grade Gliomas. J. Neurosurg. 2018, 128, 1091–1101.
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.; Gainche, L.; Sena-Esteves, M.; Curry, W.T.; Carter, R.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma Microvesicles Transport RNA and Protein That Promote Tumor Growth and Provide Diagnostic Biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476.

