Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | sami khabthani | + 1702 word(s) | 1702 | 2021-11-08 02:40:14 | | | |
| 2 | Bruce Ren | Meta information modification | 1702 | 2021-11-15 01:47:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Khabthani, S. Nonribosomal Peptide and Polyketide Against Human Pathogens. Encyclopedia. Available online: https://encyclopedia.pub/entry/15955 (accessed on 22 July 2026).
Khabthani S. Nonribosomal Peptide and Polyketide Against Human Pathogens. Encyclopedia. Available at: https://encyclopedia.pub/entry/15955. Accessed July 22, 2026.
Khabthani, Sami. "Nonribosomal Peptide and Polyketide Against Human Pathogens" Encyclopedia, https://encyclopedia.pub/entry/15955 (accessed July 22, 2026).
Khabthani, S. (2021, November 12). Nonribosomal Peptide and Polyketide Against Human Pathogens. In Encyclopedia. https://encyclopedia.pub/entry/15955
Khabthani, Sami. "Nonribosomal Peptide and Polyketide Against Human Pathogens." Encyclopedia. Web. 12 November, 2021.
Copy Citation
Antibiotics are majorly important molecules for human health. Following the golden age of antibiotic discovery, a period of decline ensued, characterised by the rediscovery of the same molecules. At the same time, new culture techniques and high-throughput sequencing enabled the discovery of new microorganisms that represent a potential source of interesting new antimicrobial substances to explore.
new NRP-PK antibiotics
in silico/in vitro strategies
human pathogens
bottom-up approach
1. Introduction and Historical Context
Antimicrobials are probably one of the most successful forms of medical treatments in the history of medicine. Their discovery is due both to chance and to the ingenuity of Alexander Fleming, who noticed that Penicillium notatum inhibited the growth of Staphylococcus aureus colonies. This observation gave rise to the antibiotic era through the discovery of penicillin. Penicillin was purified and isolated, but not industrialised at this point [1]. Industrial production came over 10 years later, led by Howard Florey and Ernst Boris Chain, using Penicillium chrysogenum [2]. René Dubos, a French microbiologist, believed in the principle of the soil providing natural active substances against pathogenic bacteria. This was based on his experiments on “antibiosis”. In 1939, Dubos discovered gramicidin, the first clinically used antibiotic [3]. This experimental design inspired researchers to develop new methods to discover antibiotics. Waksman established a platform that consists of screening mainly soil-derived bacteria, particularly Actinomycetes [4], and identified actinomycin and streptomycin. These efforts opened the door to the “golden age of antibiotic discovery” [5]. Pharmaceutical companies have continued to apply these approaches to extract and purify most of the antibiotics used today, such as erythromycin, tetracycline, vancomycin, rifamycin, and others [6]. In the 1960s, the rate of discovery of new antibiotics dropped sharply due to the high rate of rediscovery and the difficulties of characterising unknown compounds. This allowed the pharmaceutical companies to turn away from this type of research [7][8]. In the past fifty decades, only two new classes of antibiotics have been discovered. With this, international organisations have noticed a serious problem, now considering antimicrobial resistance as a major public health problem [9].
The use of natural antimicrobial compounds in human treatment is a great example of a diversion of developing resources from microbes. These secondary metabolites have been reengineered to be used by humans in order to combat several infectious diseases. They had a positive impact on human health, and they helped to stem the scourge of several diseases. Fundamentally, however, bacteria use these compounds for their self-defence. They evolve in complex ecosystems in which they are continuously in “war with one another” to ensure their own survival. To this end, and coupled with other strategies, they release antimicrobial substances into the environment. Then, they consequently contribute to the regulation of the populations of other bacterial populations with which they compete [10][11]. A number of marketed antibiotics are nonribosomal peptides (NRP) and polyketides (PK), which have been extracted from microbes in culture media, and have sometimes been modified to have a better efficiency or to reduce toxicity. It should be noted that the total pharmaceutical synthesis of these compounds is severely limited by the singularity and specificity of their ribosomally independent natural synthesis process. NRP and PK are synthetised on large nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) enzyme complexes, respectively. These enzymes are modular enzymes that function in an assembly line fashion [12][13]. These mega-enzymes are encoded in the bacterial genome by biosynthetic gene cluster (BGCs) [14].
Because of the antimicrobial properties of NRPS and PKS products, much effort has gone in to the exploration of novel NRP and PK, with the aim of developing new approaches to fight the emerging resistance profile of pathogenic bacteria. With the improvement of new sequencing technologies and bioinformatic software [15], genome mining is becoming a key strategy to discover new antibiotics. This is due to its ability to easily screen for interesting bacterial genomes and metagenomes at a constantly decreasing cost and with better efficiency.
2. Overview of Polyketide Synthase (PKS) and Nonribosomal Peptide Synthase (NRPS)
NRP and PK are two diverse families with a broad variety of complex chemical structures and pharmacological activities [13]. A large proportion of the antibiotics used in human medicine belong to the NRP and PK classes, and they are well known in infectiology, for example penicillin, vancomycin, daptomycin, erythromycin, mupirocin, and oxytetracycline (Table 1). Since the first observations were made by the chemist Jamie Collie at the University of London in 1893, establishing the structure of orcinol, to the theory of Robert Robinson in 1955 and Birch’s work, many attempts have been made to characterise the biosynthetic pathways of these secondary metabolites [12]. The multienzymatic thiotemplate model was retained as a plausible explanation, and a growing number of enzymatic domains have been identified. These domains are involved in a variety of reactions necessary for the basic assembly line system.
Table 1. Nonribosomal peptide (NRP) and polyketide (PK) molecules used currently in human medicine.
Inspired by the study of the biosynthesis of actinorhodin, [16] researchers identified the erythromycin BGC using different strategies, including sequencing adjacent parts of the gene coding for erythromycin resistance [17], looking for sequences similar to fatty acid sequence and other PKS enzymes [17], or mutated genes involved in the synthesis of 6-deoxyerythronolide B (6-dEB) [18][19]. Erythromycin polyketide synthase is encoded by three genes, eryAI, eryAII, and eryAIII, which code for three multienzymatic polypeptide 6-deoxyerythronolide B synthases, DEBS1, DEBS2, and DEBS3, respectively. Each of these giant proteins contain domains or catalytic sites [12]. Erythromycin is synthetised according to the biosynthesis mode of type I PKS. Type I PKS is a multifunctional enzyme organised into several modules (Figure 1). Each module contains three core domains necessary for the definition of type 1 PKS, namely acyl transferase (AT), ketosynthase (KS), and acyl carrier protein (ACP). The biosynthesis mode of type I PKS is linear. An acyl-coenzyme A is used as substrate and is selected by the AT. ACP, then, transfers the acyl-coenzyme A into the next module, and KS catalyses a Claisen condensation between acyl-coenzyme A and the growing polyketide chain attached to the ACP domain (Figure 1). Recent studies have questioned the definition of modules in polyketide synthase based on evolutionary analysis [20]. The authors show that domains that migrate together over the course of evolution of PKS assembly lines do not correspond to the known definitions of modules [21]. Two other types of PKS biosynthesis are known: type II and type III PKS [22]. The type II PKS is iterative; it is composed of two core domains: heterodimeric KS (KS and chain length factor subunits) and an ACP. Type II PKS usually acts by loading an α-carboxylated precursor onto an ACP, which is transferred to the active site of a KS for Claisen condensation and iterative lengthening using malonyl-coenzyme A (CoA) to result in a poly-β-keto chain [23]. This enzymatic machinery is responsible for the synthesis of aromatic polyketide such as tetracycline. Type III PKS are ACP independent and iterative homodimeric enzymes, composed of a single domain KS that plays the main role in the biosynthesis of natural products [24]. Type III PKSs are structurally less complex than type I or II PKSs and at least 15 families of type III PKSs have been identified to date [25].
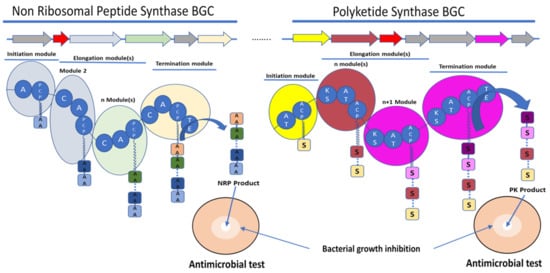
Figure 1. Schematic representation of the biosynthetic gene clusters (BGC) (arrows) and their encoded synthetase assembly lines (similarly coloured bubbles) involved in the biosynthesis of non-ribosomal peptide (NRP) and polyketide (PK) molecules. Non-ribosomal peptide synthase (NRPS) core domains are labelled as follow: A—adenylation domain, C—condensation domain, and PCP—peptidyl carrier protein. Polyketide synthase (PKS) core domains are labelled as follow: AT—acetylation domain, KS—ketosynthase, and ACP—acyl carrier protein. TE—thioesterase was a common domain to NRPS and PKS. These domains have various functions in the synthesis of the final molecule. A and AT domains are involved in the selection and activation of the substrate, C and KS domains are involved in the condensation of the substrate AA for amino acid or S for acyl-CoA or malonyl-CoA with the growing NRP or PK, respectively. ACP and PCP play a role in the transfer of the substrate between the different modules. TE releases the final molecule. The red arrows rather encode for immunity or resistance genes to the synthesized antibiotic.
The ribosome-independent synthesis mechanism of NRPS was discovered by studying the biosynthesis of unusual peptides in the Bacillus species [13]. NRPS are multienzyme machineries that assemble peptides with diverse structures and functionalities [26]. Therefore, the amino acid substrate is activated by adenylation and thioesterification at specific reactive thiol groups, the peripheral cysteines of the multienzyme, on the same model of the fatty acid synthase. An intrinsic 4′-phosphopantetheine carrier was considered to interact with the thioesterified amino acids, ensuring a step-by-step elongation of the peptide product in a series of transpeptidation and transthiolation reactions. Therefore, NRPS are organised into distinct modular sections, each composed of catalytic domains with different functions. Three domains are considered as core and are essential for peptide elongation: adenylation (A) domains, peptidyl carrier protein or thiolation (PCP or T) domains, and condensation (C) domains [27]. The A domain is involved in the selection and activation of amino acid substrates with adenosine-5′-triphosphate (ATP), and it ligates it to the adjacent PCP domain to be incorporated into the NRP [28]. A domains are, therefore, involved in the large diversity of the NRP structure by selecting not only the 20 standard proteinogenic amino acids but also nonproteinogenic amino acids [13][29]. A domains own a specific code composed of 10 amino acids, which are responsible for substrate binding [30]. Some bioinformatics tools are able to predict the substrate to be selected by an amino acid by A domains and, therefore, the NRP amino acid composition [31][32][33]. C domains catalyse amide bond formation between amino acids and the growing peptide; they have a conserved HHxxxDG motif. This conserved motif is essential for the peptide bond formation [34]. Bioinformatic analyses revealed subclasses that are classified depending on the condensation between acceptor and donor and noncanonical members of the C superfamily [35]. The PCP domains transport the substrate between the different NRPS modules [35]. PCP domains must be post-translationally modified by the addition of the 4′-phosphopantetheine (PPT) to a conserved serin residue of the conserved GGXS motif [35]. The PCP domain does not play an active role in catalysis, but it has a central role in NRPS function. The PCP interacts with the A domain and other catalytic domains involved in the biosynthesis of NRP (Figure 1) [36].
References
- Fleming, A. Classics in infectious diseases: On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae by Alexander Fleming, Reprinted from the British Journal of Experimental Pathology 10:226-236, 1929. Rev. Infect. Dis. 1980, 2, 129–139.
- Gaynes, R. The Discovery of Penicillin—New Insights after More Than 75 Years of Clinical Use. Emerg. Infect. Dis. 2017, 23, 849–853.
- Van Epps, H.L. René Dubos: Unearthing antibiotics. J. Exp. Med. 2006, 203, 259.
- Valiquette, L.; Laupland, K.B. Digging for New Solutions. Can. J. Infect. Dis. Med. Microbiol. 2015, 26, 289–290.
- Lyddiard, D.; Jones, G.L.; Greatrex, B.W. Keeping it simple: Lessons from the golden era of antibiotic discovery. FEMS Microbiol. Lett. 2016, 363, fnw084.
- Mahajan, G.B. Antibacterial Agents from Actinomycetes—A Review. Front. Biosci. 2012, 4, 240–253.
- Lewis, K. Recover the lost art of drug discovery. Nature 2012, 485, 439–440.
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387.
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic discovery: History, methods and perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382.
- Pishchany, G.; Kolter, R. On the possible ecological roles of antimicrobials. Mol. Microbiol. 2020, 113, 580–587.
- Van Bergeijk, D.A.; Terlouw, B.R.; Medema, M.H.; Van Wezel, G.P. Ecology and genomics of Actinobacteria: New concepts for natural product discovery. Nat. Rev. Genet. 2020, 18, 1–13.
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416.
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevels, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal Peptide Synthetases Involved in the Production of Medically Relevant Natural Products. Mol. Pharm. 2008, 5, 191–211.
- Walsh, C.T.; Fischbach, M.A. Natural Products Version 2.0: Connecting Genes to Molecules. J. Am. Chem. Soc. 2010, 132, 2469–2493.
- Tracanna, V.; De Jong, A.; Medema, M.H.; Kuipers, O.P. Mining prokaryotes for antimicrobial compounds: From diversity to function. FEMS Microbiol. Rev. 2017, 41, 417–429.
- Malpartida, F.; Hopwood, D.A. Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature 1984, 309, 462–464.
- Cortes, J.; Haydock, S.F.; Roberts, G.A.; Bevitt, D.J.; Leadlay, P. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 1990, 348, 176–178.
- Stanzak, R.; Matsushima, P.; Baltz, R.H.; Rao, R.N. Cloning and Expression in Streptomyces lividans of Clustered Erythromycin Biosynthesis Genes from Streptomyces erythreus. Nat. Biotechnol. 1986, 4, 229–232.
- Weber, J.; Losick, R. The use of a chromosome integration vector to map erythromycin resistance and production genes in Saccharopolyspora erythraea (Streptomyces erythraeus). Gene 1988, 68, 173–180.
- Keatinge-Clay, A.T. Polyketide Synthase Modules Redefined. Angew. Chem. Int. Ed. 2017, 56, 4658–4660.
- Zhang, L.; Hashimoto, T.; Qin, B.; Hashimoto, J.; Kozone, I.; Kawahara, T.; Okada, M.; Awakawa, T.; Ito, T.; Asakawa, Y.; et al. Characterization of Giant Modular PKSs Provides Insight into Genetic Mechanism for Structural Diversification of Aminopolyol Polyketides. Angew. Chem. Int. Ed. 2017, 56, 1740–1745.
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295.
- Zhang, Z.; Pan, H.-X.; Tang, G.-L. New insights into bacterial type II polyketide biosynthesis. F1000Research 2017, 6, 172.
- Shimizu, Y.; Ogata, H.; Goto, S. Type III Polyketide Synthases: Functional Classification and Phylogenomics. ChemBioChem 2016, 18, 50–65.
- Lim, Y.P.; Go, M.K.; Yew, W.S. Exploiting the Biosynthetic Potential of Type III Polyketide Synthases. Molecules 2016, 21, 806.
- Süssmuth, R.D.; Mainz, A. Nonribosomal Peptide Synthesis-Principles and Prospects. Angew. Chem. Int. Ed. 2017, 56, 3770–3821.
- Strieker, M.; Tanović, A.; Marahiel, M.A. Nonribosomal peptide synthetases: Structures and dynamics. Curr. Opin. Struct. Biol. 2010, 20, 234–240.
- Stanišić, A.; Kries, H. Adenylation Domains in Nonribosomal Peptide Engineering. ChemBioChem 2019, 20, 1347–1356.
- Hancock, R.E.W.; Chapple, D.S. Peptide Antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323.
- Stachelhaus, T.; Mootz, H.D.; Marahiel, M.A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999, 6, 493–505.
- Prieto, C.; Garcia-Estrada, C.; Lorenzana, D.; Martín, J.F. NRPSsp: Non-ribosomal peptide synthase substrate predictor. Bioinformatics 2011, 28, 426–427.
- Lee, T.V.; Johnson, R.D.; Arcus, V.L.; Lott, J.S. Prediction of the substrate for nonribosomal peptide synthetase (NRPS) adenylation domains by virtual screening. Proteins Struct. Funct. Bioinform. 2015, 83, 2052–2066.
- Chevrette, M.G.; Aicheler, F.; Kohlbacher, O.; Currie, C.R.; Medema, M.H. SANDPUMA: Ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 2017, 33, 3202–3210.
- Stachelhaus, T.; Mootz, H.D.; Bergendahl, V.; Marahiel, M.A. Peptide Bond Formation in Nonribosomal Peptide Biosynthesis. J. Biol. Chem. 1998, 273, 22773–22781.
- Bloudoff, K.; Schmeing, T.M. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2017, 1865, 1587–1604.
- Owen, J.G.; Calcott, M.J.; Robins, K.J.; Ackerley, D.F. Generating Functional Recombinant NRPS Enzymes in the Laboratory Setting via Peptidyl Carrier Protein Engineering. Cell Chem. Biol. 2016, 23, 1395–1406.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Peptides for Health Benefits
Revisions:
2 times
(View History)
Update Date:
15 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No