 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rebecca Dauwe | + 3985 word(s) | 3985 | 2021-11-08 02:12:42 | | | |
| 2 | Bruce Ren | Meta information modification | 3985 | 2021-11-15 01:45:18 | | |
Video Upload Options
The plant specialized metabolome consists of a multitude of structurally and functionally diverse metabolites, variable from species to species. The specialized metabolites play roles in the response to environmental changes and abiotic or biotic stresses, as well as in plant growth and development. At its basis, the specialized metabolism is built of four major pathways, each starting from a few distinct primary metabolism precursors, and leading to distinct basic carbon skeleton core structures: polyketides and fatty acid derivatives, terpenoids, alkaloids, and phenolics. Structural diversity in the specialized metabolism further expands exponentially with each subsequent modification of the core structure, decoration and conjugation. Series of subsequent conjugations among decorated specialized metabolites lead to the formation of polyglycosides and polyesters in rare cases of macromolecular weight. Many specialized metabolites are involved in redox reactions, and part of the structural diversity can also be attributed to follow-up reactions among oxidized structures, leading to the extremely diverse array of biflavonoids, (neo)lignans, oligolignols, proanthocyanidines and phlobaphenes, and derivatives of aromatically decorated compounds.
1. The Four Major Specialized Metabolome Pathways
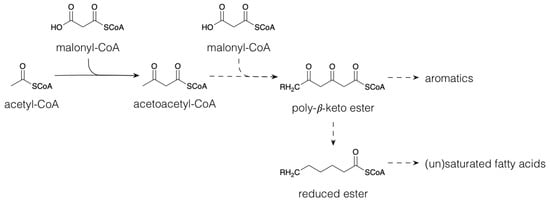
1.1. Polyketides and Fatty Acid Derivatives
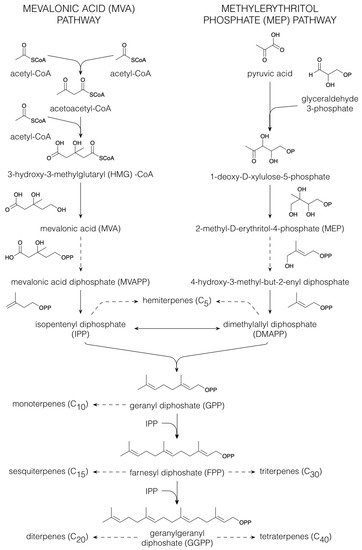
1.2. Terpenoids
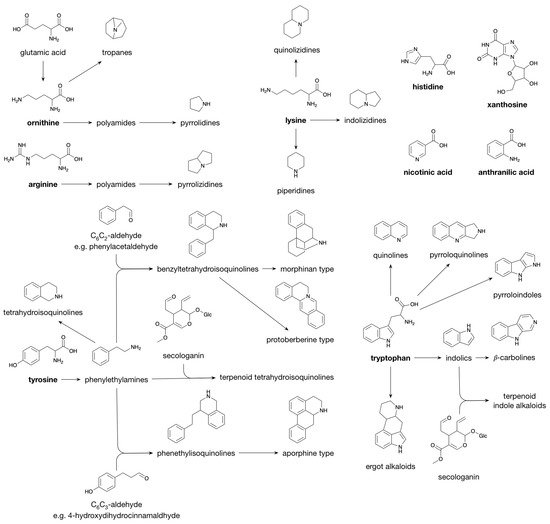
1.3. Alkaloids
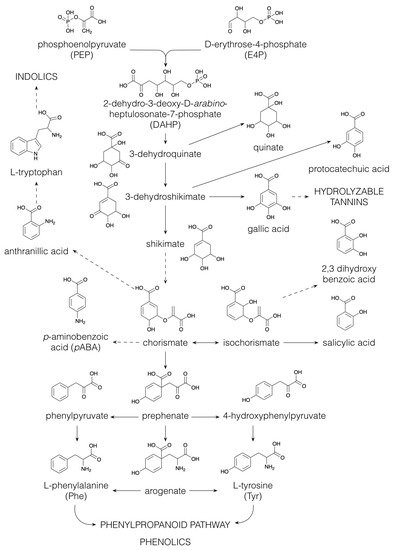
1.4. Phenolics
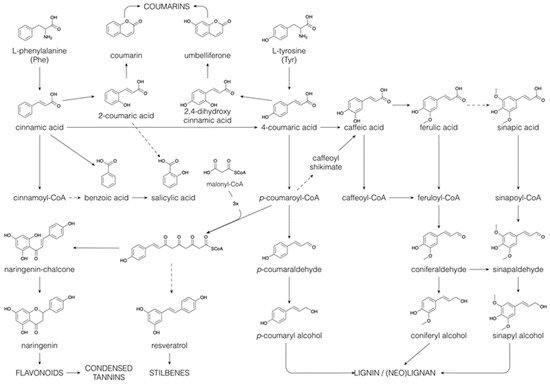
2. Decoration and Conjugation in Specialized Metabolism
2.1. Diversity of Decorations
The majority of the structural diversity in the plant specialized metabolome arises from “decorative” biosynthetic steps, such as hydroxylations, methylations, glycosylations, acylations and prenylations [81]. The number of different modifications is rather limited and the nature of the decorations for different classes of specialized metabolites is often the same. For example, classes as far apart as flavonoids, indole alkaloids and apocarotenoids are all decorated with glucose and with malonylglucose [82][83]. In the specialized metabolite profile of a plant extract, the common decorations are reflected in an overrepresentation of certain mass differences corresponding to the difference between two core structures. For example, in the maize specialized metabolome, the mass difference of 108.021 Da, corresponding to the difference between a phenylpropanoid and a flavonoid core structure, is observed between p-coumaric acid and naringenin, but also between caffeoyl hexose and eriodictyol 7-O-hexoside (hydroxylation and addition of a hexose to both) or between sinapoyl hexose and methoxy-homoeriodictyol (two methoxylations of both), as well as between dihydroxyindole-3-acetic acid (caffeoyl) hexoside and dihydroxyindole-3-acetic acid (eriodictyol-O-) hexoside (hydroxylation and addition of dihydroxyindole-3-acetic acid and hexose to both) [33]. The majority of plant specialized metabolites are glycosylated and/or acylated, with acylations being aromatic (p-coumaric, caffeic, ferulic, sinapic, gallic or p-hydroxybenzoic acids) or aliphatic (malonic, acetic, malic, succinic or oxalic acids). Because of the multitude of different possible decoration sites (hydroxyl groups), and the multitude of possible combinations of decorations, the number of possible structures grows exponentially with each added decoration [84]. Moreover, N-acylation of alkaloids, such as anthranilic acid or polyamines, leads to the formation of a high variety of hydroxycinnamic acid amides [85]. Intramolecular cyclization through phenol-oxidative coupling between the phenolic rings of two p-coumaroyl substitutes in bis-(p-coumaroyl) polyamines is the biosynthetic mechanism leading to cyclic alkaloids such as lunarine (in Lunaria annua seeds) and aphelandrine (in Aphelandra sp. roots) [86][87]. The introduction of phenolic rings through aromatic acylation allows, thus, further structural diversification through oxidative coupling.
2.2. High Molecular Weight Conjugates
Glucosylation and acylation allow for the concatenation of several specialized metabolites, sometimes of different metabolic classes. In the maize specialized metabolome, phenylpropanoid glycosides are coupled to benzoic acids, flavonoids, phenylethanoids and indolics [33]. Hexosylated flavonoids, but also free hexoses or dihexoses, often carry multiple aromatic acylations, often of different natures [83][33][88]. Less frequently, the hexose moiety in poly-acylated aromatic conjugates is replaced by an alternative polyol, such as glucaric acid [89]. The feruloyl moieties, both in glucaric acid and hexose conjugates, are sometimes coupled by radical–radical coupling to mono- or oligolignols, allowing these coupling products to be stored as such in the leaf vacuole [89][54]. In contrast to glucaric acid conjugates, where the two carboxylic acid groups of glucaric acid remain unesterified, other aromatic conjugates involve esterification to the carboxyl groups of dicarboxylic acids such as succinic acid or 3-hydroxy-3-methylglutaric acid. For example, in blue Agapanthus flowers, a p-coumaroylated delphinidin diglycoside is attached to a favonol triglycoside via a succinic acid diester link [90]. In this conjugate, the aromatic acyl group and the colourless flavonoid both stabilize the blue color of the anthocyanin delphidin diglycoside. Esterification involving the dicarboxylic acids 3-hydroxy-3-methylglutaric acid (HMGA) and succinic acid allowed the formation of a macromolecular polyester structure carrying glucosylated lignans, glucosylated flavonoids, phenylpropanoic acids and ferulic acid coupled oligolignols in the outer integument of flax seeds [84]. More common polyesters in higher plants are cutin and suberin, of which the latter contains aromatic domains derived from cinnamic acids [91]. Cutin and suberin function as protecting barriers against desiccation or biotic and abiotic stresses.
References
- Dewick, P.M. The acetate pathway: Fatty acids and polyketides. In Medicinal Natural Products; John Wiley & Sons Ltd: Hoboken, NJ, USA, 2009; pp. 39–135.
- Flores-Sanchez, I.J.; Verpoorte, R. Plant polyketide synthases: A fascinating group of enzymes. Plant Physiol. Biochem. PPB 2009, 47, 167–174.
- Blée, E. Phytooxylipins and plant defense reactions. Prog. Lipid Res. 1998, 37, 33–72.
- Vincenti, S.; Mariani, M.; Alberti, J.-C.; Jacopini, S.; De Caraffa, V.B.-B.; Berti, L.; Maury, J. Biocatalytic Synthesis of Natural Green Leaf Volatiles Using the Lipoxygenase Metabolic Pathway. Catalysts 2019, 9, 873.
- Wasternack, C. Jasmonates: An Update on Biosynthesis, Signal Transduction and Action in Plant Stress Response, Growth and Development. Ann. Bot. 2007, 100, 681–697.
- Wasternack, C.; Hause, B. Jasmonates: Biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in Annals of Botany. Ann. Bot. 2013, 111, 1021–1058.
- Matsui, K. Green leaf volatiles: Hydroperoxide lyase pathway of oxylipin metabolism. Curr. Opin. Plant Biol. 2006, 9, 274–280.
- Matsui, K.; Koeduka, T. Green leaf volatiles in plant signaling and response. In Lipids in Plant and Algae Development; Nakamura, Y., Li-Beisson, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 427–443.
- Frost, C.J.; Mescher, M.C.; Dervinis, C.; Davis, J.M.; Carlson, J.E.; De Moraes, C.M. Priming defense genes and metabolites in hybrid poplar by the green leaf volatile cis-3-hexenyl acetate. New Phytol. 2008, 180, 722–734.
- McCormick, A.C.; Irmisch, S.; Reinecke, A.; Boeckler, G.A.; Veit, D.; Reichelt, M.; Hansson, B.S.; Gershenzon, J.; Köllner, T.; Unsicker, S.B. Herbivore-induced volatile emission in black poplar: Regulation and role in attracting herbivore enemies. Plant Cell Environ. 2014, 37, 1909–1923.
- Scala, A.; Allmann, S.; Mirabella, R.; Haring, M.A.; Schuurink, R.C. Green Leaf Volatiles: A Plant’s Multifunctional Weapon against Herbivores and Pathogens. Int. J. Mol. Sci. 2013, 14, 17781–17811.
- Zwenger, S.R.; Basu, C. Plant terpenoids: Applications and future potentials. Biotechnol. Mol. Biol. Rev. 2008, 3, 1–7.
- Zi, J.; Mafu, S.; Peters, R.J. To gibberellins and beyond! Surveying the evolution of (di)terpenoid metabolism. Annu. Rev. Plant Biol. 2014, 65, 259–286.
- Pichersky, E.; Raguso, R.A. Why do plants produce so many terpenoid compounds? New Phytol. 2018, 220, 692–702.
- Dewick, P.M. The mevalonate and methylerythritol phosphate pathways: Terpenoids and steroids. In Medicinal Natural Products; John Wiley & Sons Ltd: Hoboken, NJ, USA, 2009; pp. 187–310.
- Lipko, A.; Swiezewska, E. Isoprenoid generating systems in plants—A handy toolbox how to assess contribution of the mevalonate and methylerythritol phosphate pathways to the biosynthetic process. Prog. Lipid Res. 2016, 63, 70–92.
- Bohlmann, J.; Keeling, C.I. Terpenoid biomaterials. Plant J. Cell Mol. Biol. 2008, 54 4, 656–669.
- Vranová, E.; Coman, D.; Gruissem, W. Structure and dynamics of the isoprenoid pathway network. Mol. Plant 2012, 5, 318–333.
- Loreto, F.; Dicke, M.; Schnitzler, J.-P.; Turlings, T.C.J. Plant volatiles and the environment. Plant Cell Environ. 2014, 37, 1905–1908.
- Tholl, D. Biosynthesis and biological functions of terpenoids in plants. Adv. Biochem. Eng. Biotechnol. 2015, 148, 63–106.
- Gershenzon, J.; Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007, 3, 408–414.
- Block, A.K.; Vaughan, M.M.; Schmelz, E.A.; Christensen, S.A. Biosynthesis and function of terpenoid defense compounds in maize (Zea mays). Planta 2019, 249, 21–30.
- Veenstra, A.; Moola, N.; Wighard, S.S.; Korsman, J.; Christensen, S.A.; Rafudeen, M.S.; Murray, S.L. Kauralexins and zealexins accumulate in sub-tropical maize lines and play a role in seedling resistance to Fusarium verticillioides. Eur. J. Plant Pathol. 2018, 153, 223–237.
- Rasmann, S.; Köllner, T.G.; Degenhardt, J.; Hiltpold, I.; Toepfer, S.; Kuhlmann, U.; Gershenzon, J.; Turlings, T.C.J. Recruitment of entomopathogenic nematodes by insect-damaged maize roots. Nature 2005, 434, 732–737.
- Irmisch, S.; Jiang, Y.; Chen, F.; Gershenzon, J.; Köllner, T.G. Terpene synthases and their contribution to herbivore-induced volatile emission in western balsam poplar (Populus trichocarpa). BMC Plant Biol. 2014, 14, 1–16.
- Lackus, N.D.; Lackner, S.; Gershenzon, J.; Unsicker, S.; Köllner, T.G. The occurrence and formation of monoterpenes in herbivore-damaged poplar roots. Sci. Rep. 2018, 8, 17936.
- Dewick, P.M. Alkaloids. In Medicinal Natural Products; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 311–420.
- Chen, D.; Shao, Q.; Yin, L.; Younis, A.; Zheng, B. Polyamine Function in Plants: Metabolism, Regulation on Development, and Roles in Abiotic Stress Responses. Front. Plant Sci. 2019, 9, 1945.
- Azzeme, A.; Kamarul Zaman, M. Plant toxins: Alkaloids and their toxicities. GSC Biol. Pharm. Sci. 2019, 6, 21–29.
- Matsuura, H.; Fett-Neto, A. Plant alkaloids: Main features, toxicity, and mechanisms of action. Plant Toxins 2017, 2, 243–261.
- Feng, G.; Chen, M.; Ye, H.-C.; Zhang, Z.-K.; Li, H.; Chen, L.-L.; Chen, X.-L.; Yan, C.; Zhang, J. Herbicidal activities of compounds isolated from the medicinal plant Piper sarmentosum. Ind. Crop. Prod. 2019, 132, 41–47.
- Debnath, B.; Singh, W.S.; Das, M.; Goswami, S.; Singh, M.K.; Maiti, D.; Manna, K. Role of plant alkaloids on human health: A review of biological activities. Mater. Today Chem. 2018, 9, 56–72.
- Desmet, S.; Saeys, Y.; Verstaen, K.; Dauwe, R.; Kim, H.; Niculaes, C.; Fukushima, A.; Goeminne, G.; Vanholme, R.; Ralph, J.; et al. Maize specialized metabolome networks reveal organ-preferential mixed glycosides. Comput. Struct. Biotechnol. J. 2021, 19, 1127–1144.
- Zhou, S.; Richter, A.; Jander, G. Beyond defense: Multiple functions of benzoxazinoids in maize metabolism. Plant Cell Physiol. 2018, 59, 1528–1537.
- Niemeyer, H.M. Hydroxamic acids derived from 2-hydroxy-2H-1,4-benzoxazin-3(4H)-one: Key defense chemicals of cereals. J. Agric. Food Chem. 2009, 57, 1677–1696.
- Oikawa, A.; Ishihara, A.; Tanaka, C.; Mori, N.; Tsuda, M.; Iwamura, H. Accumulation of HDMBOA-Glc is induced by biotic stresses prior to the release of MBOA in maize leaves. Phytochemistry 2004, 65, 2995–3001.
- Cheynier, V.; Comte, G.; Davies, K.M.; Lattanzio, V.; Martens, S. Plant phenolics: Recent advances on their biosynthesis, genetics, and ecophysiology. Plant Physiol. Biochem. PPB 2013, 72, 1–20.
- Dewick, P.M. The shikimate pathway: Aromatic amino acids and phenylpropanoids. In Medicinal Natural Products; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 137–186.
- Maeda, H.; Dudareva, N. The Shikimate Pathway and Aromatic Amino Acid Biosynthesis in Plants. Annu. Rev. Plant Biol. 2012, 63, 73–105.
- Grundhöfer, P.; Niemetz, R.; Schilling, G.; Gross, G.G. Biosynthesis and subcellular distribution of hydrolyzable tannins. Phytochemistry 2001, 57, 915–927.
- Strobbe, S.; Van Der Straeten, D. Toward Eradication of B-Vitamin Deficiencies: Considerations for Crop Biofortification. Front. Plant Sci. 2018, 9, 443.
- Strobbe, S.; Van Der Straeten, D. Folate biofortification in food crops. Curr. Opin. Biotechnol. 2017, 44, 202–211.
- Naqvi, S.; Zhu, C.; Farre, G.; Ramessar, K.; Bassie, L.; Breitenbach, J.; Conesa, D.P.; Ros, G.; Sandmann, G.; Capell, T.; et al. Transgenic multivitamin corn through biofortification of endosperm with three vitamins representing three distinct metabolic pathways. Proc. Natl. Acad. Sci. USA 2009, 106, 7762–7767.
- Gallei, M.; Luschnig, C.; Friml, J. Auxin signalling in growth: Schrödinger’s cat out of the bag. Curr. Opin. Plant Biol. 2020, 53, 43–49.
- Tzin, V.; Galili, G. New Insights into the Shikimate and Aromatic Amino Acids Biosynthesis Pathways in Plants. Mol. Plant 2010, 3, 956–972.
- Yoo, H.; Widhalm, J.R.; Qian, Y.; Maeda, H.; Cooper, B.R.; Jannasch, A.S.; Gonda, I.; Lewinsohn, E.; Rhodes, D.; Dudareva, N. An alternative pathway contributes to phenylalanine biosynthesis in plants via a cytosolic tyrosine:phenylpyruvate aminotransferase. Nat. Commun. 2013, 4, 2833.
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20.
- Widhalm, J.; Dudareva, N. A Familiar Ring to It: Biosynthesis of Plant Benzoic Acids. Mol. Plant 2015, 8, 83–97.
- Chen, Z.; Zheng, Z.; Huang, J.; Lai, Z.; Fan, B. Biosynthesis of salicylic acid in plants. Plant Signal. Behav. 2009, 4, 493–496.
- Mahdi, J.G. Biosynthesis and metabolism of β-d-salicin: A novel molecule that exerts biological function in humans and plants. Biotechnol. Rep. 2014, 4, 73–79.
- Boeckler, G.A.; Gershenzon, J.; Unsicker, S.B. Phenolic glycosides of the Salicaceae and their role as anti-herbivore defenses. Phytochemistry 2011, 72, 1497–1509.
- Vanholme, R.; De Meester, B.; Ralph, J.; Boerjan, W. Lignin biosynthesis and its integration into metabolism. Curr. Opin. Biotechnol. 2019, 56, 230–239.
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546.
- Dima, O.; Morreel, K.; Vanholme, B.; Kim, H.; Ralph, J.; Boerjan, W. Small Glycosylated Lignin Oligomers Are Stored in Arabidopsis Leaf Vacuoles. Plant Cell 2015, 27, 695–710.
- Niculaes, C.; Morreel, K.; Kim, H.; Lu, F.; McKee, L.S.; Ivens, B.; Haustraete, J.; Vanholme, B.; De Rycke, R.; Hertzberg, M.; et al. Phenylcoumaran Benzylic Ether Reductase Prevents Accumulation of Compounds Formed under Oxidative Conditions in Poplar Xylem. Plant Cell 2014, 26, 3775–3791.
- Moss, G.P. Nomenclature of Lignans and Neolignans (IUPAC Recommendations 2000). Pure Appl. Chem. 2000, 72, 1493–1523.
- Cong, L.H.; Dauwe, R.; Lequart, M.; Vinchon, S.; Renouard, S.; Fliniaux, O.; Colas, C.; Corbin, C.; Doussot, J.; Hano, C.; et al. Kinetics of glucosylated and non-glucosylated aryltetralin lignans in Linum hairy root cultures. Phytochemistry 2015, 115, 70–78.
- Jullian-Pawlicki, N.; Lequart-Pillon, M.; Huynh-Cong, L.; Lesur, D.; Cailleu, D.; Mesnard, F.; Laberche, J.C.; Gontier, E.; Boitel-Conti, M. Arylnaphthalene and aryltetralin-type lignans in hairy root cultures ofLinum perenne, and the stereochemistry of 6-methoxypodophyllotoxin and one diastereoisomer by HPLC-MS and NMR spectroscopy. Phytochem. Anal. 2015, 26, 310–319.
- Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P.F.; Marita, J.M.; Hatfield, R.D.; Ralph, S.A.; Christensen, J.H.; et al. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenyl- propanoids. Phytochem. Rev. 2004, 3, 29–60.
- Van Acker, R.; Déjardin, A.; Desmet, S.; Hoengenaert, L.; Vanholme, R.; Morreel, K.; Laurans, F.; Kim, H.; Santoro, N.; Foster, C.; et al. Different Routes for Conifer- and Sinapaldehyde and Higher Saccharification upon Deficiency in the Dehydrogenase CAD1. Plant Physiol. 2017, 175, 1018–1039.
- Vanholme, R.; Ralph, J.; Akiyama, T.; Lu, F.; Pazo, J.R.; Kim, H.; Christensen, J.H.; Van Reusel, B.; Storme, V.; De Rycke, R.; et al. Engineering traditional monolignols out of lignin by concomitant up-regulation of F5H1 and down-regulation of COMT in Arabidopsis. Plant J. 2010, 64, 885–897.
- Sundin, L.; Vanholme, R.; Geerinck, J.; Goeminne, G.; Höfer, R.; Kim, H.; Ralph, J.; Boerjan, W. Mutation of the Inducible ARABIDOPSIS THALIANA CYTOCHROME P450 REDUCTASE2 Alters Lignin Composition and Improves Saccharification. Plant Physiol. 2014, 166, 1956–1971.
- Ralph, J.; Lapierre, C.; Boerjan, W. Lignin structure and its engineering. Curr. Opin. Biotechnol. 2019, 56, 240–249.
- Davies, K.M.; Albert, N.W.; Schwinn, K.E. From landing lights to mimicry: The molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct. Plant Biol. FPB 2012, 39, 619–638.
- Khalid, M.; Rahman, S.U.; Bilal, M.; Huang, D.-F. Role of flavonoids in plant interactions with the environment and against human pathogens—A review. J. Integr. Agric. 2019, 18, 211–230.
- Pilkington, L.I.; Barker, D. Synthesis and biology of 1,4-benzodioxane lignan natural products. Nat. Prod. Rep. 2015, 32, 1369–1388.
- Nyiredy, S.; Samu, Z.; Szücs, Z.; Gulácsi, K.; Kurtán, T.; Antus, S. New insight into the biosynthesis of flavanolignans in the white-flowered variant of Silybum marianum. J. Chromatogr. Sci. 2008, 46, 93–96.
- Ferreira, D.; Nel, R.J.J.; Bekker, R. 3.19-Condensed Tannins. In Comprehensive Natural Products Chemistry; Barton, S.D., Nakanishi, K., Meth-Cohn, O., Eds.; Pergamon: Oxford, UK, 1999; pp. 747–797.
- Weng, J.-K.; Noel, J.P. Chemodiversity in Selaginella: A reference system for parallel and convergent metabolic evolution in terrestrial plants. Front. Plant Sci. 2013, 4, 119.
- Andrade, A.W.L.; Machado, K.D.C.; Machado, K.D.C.; Figueiredo, D.D.R.; David, J.M.; Islam, M.T.; Uddin, S.J.; Shilpi, J.A.; Costa, J.P. In vitro antioxidant properties of the biflavonoid agathisflavone. Chem. Central J. 2018, 12, 75.
- Gontijo, V.S.; Dos Santos, M.H.; Viegas, C., Jr. Biological and chemical aspects of natural biflavonoids from plants: A brief review. Mini Rev. Med. Chem. 2017, 17, 834–862.
- Geiger, H.; Seeger, T. Triflavones and a biflavone from the moss Rhizogonium Distichum. Z. Naturforschung. 2000, 55, 870–873.
- Carneiro, F.J.; Boralle, N.; Silva, D.H.; Lopes, L.M. Bi- and tetraflavonoids from Aristolochia ridicula. Phytochemistry 2000, 55, 823–832.
- Goossens, J.-F.; Goossens, L.; Bailly, C. Hinokiflavone and Related C–O–C-Type Biflavonoids as Anti-cancer Compounds: Properties and Mechanism of Action. Nat. Prod. Bioprospecting 2021, 11, 365–377.
- Waterman, M.J.; Nugraha, A.S.; Hendra, R.; Ball, G.E.; Robinson, S.A.; Keller, P.A. Antarctic moss biflavonoids show high antioxidant and ultraviolet-screening activity. J. Nat. Prod. 2017, 80, 2224–2231.
- Ye, Y.; Guo, Y.; Luo, Y.T.; Wang, Y.F. Isolation and free radical scavenging activities of a novel biflavonoid from the shells of Camellia oleifera Abel. Fitoterapia 2012, 83, 1585–1589.
- Menezes, J.; Diederich, M.F. Bioactivity of natural biflavonoids in metabolism-related disease and cancer therapies. Pharmacol. Res. 2021, 167, 105525.
- Silva, G.L.; Chai, H.; Gupta, M.P.; Farnsworth, N.R.; Cordell, G.A.; Pezzuto, J.M.; Beecher, C.W.W.; Douglas Kinghorn, A. Cytotoxic biflavonoids from Selaginella willdenowii. Phytochemistry 1995, 40, 129–134.
- Effenberger, I.; Zhang, B.; Li, L.; Wang, Q.; Liu, Y.; Klaiber, I.; Pfannstiel, J.; Wang, Q.; Schaller, A. Dirigent proteins from cotton (Gossypium sp.) for the atropselective synthesis of gossypol. Angew. Chem. Int. Ed. 2015, 54, 14660–14663.
- Paniagua, C.; Bilkova, A.; Jackson, P.; Dabravolski, S.; Riber, W.; Didi, V.; Houser, J.; Gigli-Bisceglia, N.; Wimmerova, M.; Budínská, E.; et al. Dirigent proteins in plants: Modulating cell wall metabolism during abiotic and biotic stress exposure. J. Exp. Bot. 2017, 68, 3287–3301.
- Wang, S.; Alseekh, S.; Fernie, A.R.; Luo, J. The structure and function of major plant metabolite modifications. Mol. Plant. 2019, 12, 899–919.
- Nguyen, T.-K.; Marcelo, P.; Gontier, E.; Dauwe, R. Metabolic markers for the yield of lipophilic indole alkaloids in dried woad leaves (Isatis tinctoria L.). Phytochemistry 2019, 163, 89–98.
- Morreel, K.; Saeys, Y.; Dima, O.; Lu, F.; Van de Peer, Y.; Vanholme, R.; Ralph, J.; Vanholme, B.; Boerjan, W. Systematic Structural Characterization of Metabolites in Arabidopsis via Candidate Substrate-Product Pair Networks. Plant Cell 2014, 26, 929–945.
- Thiombiano, B.; Gontier, E.; Molinié, R.; Marcelo, P.; Mesnard, F.; Dauwe, R. An untargeted liquid chromatography–mass spectrometry-based workflow for the structural characterization of plant polyesters. Plant J. 2020, 102, 1323–1339.
- Luo, J.; Fuell, C.; Parr, A.; Hill, L.; Bailey, P.; Elliott, K.; Fairhurst, S.A.; Martin, C.; Michael, A.J. A Novel Polyamine Acyltransferase Responsible for the Accumulation of Spermidine Conjugates in Arabidopsis Seed. Plant Cell 2009, 21, 318–333.
- Sagner, S.; Shen, Z.; Deus-Neumann, B.; Zenk, M.H. The biosynthesis of lunarine in seeds of Lunaria annua. Phytochemistry 1998, 47, 375–387.
- Nezbedová, L.; Hesse, M.; Drandarov, K.; Bigler, L.; Werner, C. Phenol oxidative coupling in the biogenesis of the macrocyclic spermine alkaloids aphelandrine and orantine in Aphelandra sp. Planta 2001, 213, 411–417.
- Baumert, A.; Milkowski, C.; Schmidt, J.; Nimtz, M.; Wray, V.; Strack, D. Formation of a complex pattern of sinapate esters in Brassica napus seeds, catalyzed by enzymes of a serine carboxypeptidase-like acyltransferase family? Phytochemistry 2005, 66, 1334–1345.
- Nguyen, T.-K.; Jamali, A.; Grand, E.; Morreel, K.; Marcelo, P.; Gontier, E.; Dauwe, R. Phenylpropanoid profiling reveals a class of hydroxycinnamoyl glucaric acid conjugates in Isatis tinctoria leaves. Phytochemistry 2017, 144, 127–140.
- Bloor, S.J.; Falshaw, R. Covalently linked anthocyanin-flavonol pigments from blue Agapanthus flowers. Phytochemistry 2000, 53, 575–579.
- Kolattukudy, P.E. Polyesters in higher plants. Adv. Biochem. Eng. Biotechnol. 2001, 71, 1–49.

