1.2. Terpenoids
Terpenoids comprise the largest group of specialized metabolites in the plant kingdom [
30,
31,
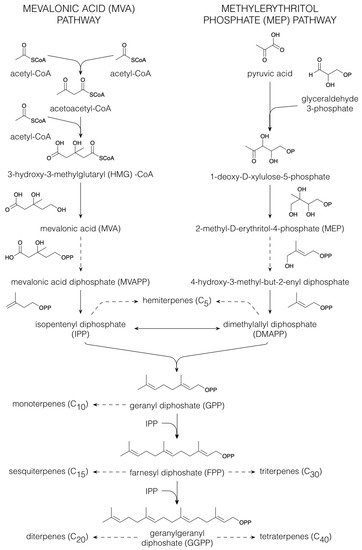
32]. All terpenoids are derived from the five-carbon building blocks, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), that are derived via either the mevalonic acid (MVA) or the plastidial 2-methyl-D-erythritol 4-phosphate (MEP) pathway [
33] (
Figure 2). In the MVA pathway, three acetyl-CoA molecules are condensed to form 3-hydroxy-3-methylglutaryl (HMG)-CoA. Following reduction in HMG-CoA to MVA, phosphorylation yields MVA diphosphate (MVAPP), from which a combined decarboxylation and dehydration results in the formation of IPP. Isomerization of IPP provides DMAPP [
33]. However, most plant terpenoids are produced via the MEP pathway [
34]. Here, decarboxylation of pyruvic acid is followed by a condensation with glyceraldehyde-3-phosphate, yielding 1-deoxy-D-xylulose-5-phosphate. The latter compound is then converted to MEP. Upon activation with cytidine triphosphate, a phosphoanhydride is formed of which the cleavage leads to 4-hydroxy-3-methyl-but-2-enyl diphosphate, which is then the immediate precursor of both IPP and DMAPP [
33]. Dephosphorylation of IPP and DMAPP yields the hemiterpenes (C
5), whereas their concatenation provides geranyl diphosphate (GPP) from which the monoterpenes (C
10) are derived. The further addition of IPP to GPP produces farnesyl diphosphate (FPP) that is central in sesquiterpene (C
15) biosynthesis. Linking another FPP to IPP yields then geranylgeranyl diphosphate (GGPP), the precursor of the diterpenes (C
20). All these coupling reactions occur via a head-to-tail condensation, yet the triterpenes (C
30) result from joining, tail-to-tail, two FPP molecules. Dependent on the compound class, tetraterpenes (C
40) are synthesized via tail-to-tail condensation of two GGPP molecules or via head-to-tail condensations [
33].
Figure 2. Biosynthesis of terpenoids. Dashed arrows indicate multiple conversions.
In plants, terpenoids play a role in plant growth and development as photosynthetic pigments (e.g., carotenoids, C
40), electron-carriers (e.g., ubiquinone, C
40), regulators of growth and development (e.g., gibberellins, C
20) or as part of membrane structures (e.g., phytosterols, C
30) [
35,
36]. In addition, volatile, low-molecular weight terpenoids (C
5-C
10) are involved in the protection of plants against biotic (e.g., microbial pathogens) and abiotic (e.g., thermal stress) stress above- and belowground [
37,
38]. Furthermore, volatile terpenoids, as constituents of floral scent, play a role in the attraction of plant pollinators [
39]. Maize produces a range of non-volatile terpenoids that are elicited in response to biotic attack [
40]. For example, phytoalexins, kauralexins and zealexins (C
15) play a role in maize seedling resistance to
Fusarium verticillioides [
41]. Furthermore, herbivory-damaged maize roots release volatile sesquiterpenes that attract entomopathogenic nematodes which are able to infest the attacking larvae [
42]. Herbivore attack of poplar leaves induced the emission of a complex mixture of volatile terpenoids (e.g., (E)-β-ocimene and (E,E)-α-farnescene) and GLVs aboveground [
43]. In addition, cockchafer larvae-damaged roots of
Populus trichocarpa and
Populus nigra release a mixture of monoterpenes, including β-pinene, camphene and 1,8-cineole [
44].
1.3. Alkaloids
Alkaloids are characterized by great structural diversity and physiological properties. The sole thing that alkaloids have in common is the presence of a nitrogen atom, mostly present in a heterocyclic ring. This nitrogen atom in alkaloids originates mainly from amino acids, and, in general, the carbon skeleton of the particular amino acid is incorporated in the alkaloid structure, hence, classification is based on their biosynthesis precursor [
45]. However, some alkaloid classes derive only their nitrogen atom from an amino acid via a transamination reaction, while the remainder of the structure is derived from, e.g., acetate, shikimate, terpenoids or steroids; these are classified as pseudoalkaloids [
45]. Most alkaloids are derived from ornithine (derived from glutamic acid in plants), arginine, lysine, tyrosine and tryptophan. Furthermore, minor alkaloid classes are produced from histidine (imidazole alkaloids), nicotinic acid (pyridine alkaloids), anthranilic acid (quinazoline alkaloids) and xanthosine (purine alkaloids) [
45].
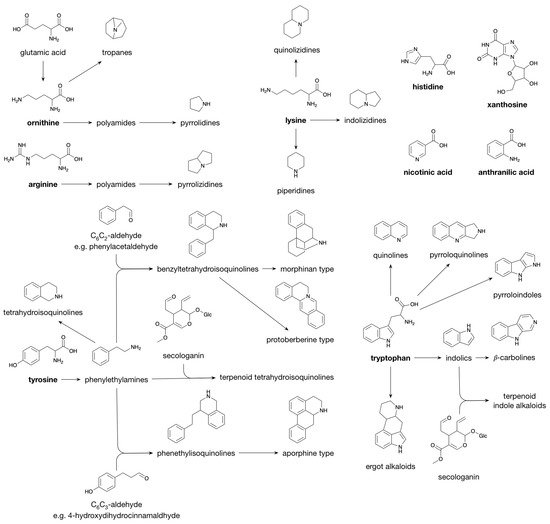
Polyamines, e.g., agmatine, putrescine and spermidine, are formed from either ornithine or arginine [
46]. Polyamines derived from ornithine yield pyrrolidines and tropanes (e.g., cocaine) upon cyclization, whereas cyclization of arginine-derived polyamines yields pyrrolizidines (
Figure 3 [
45]). Having one additional methylene moiety as compared to ornithine, lysine enters alkaloid biosynthesis pathways that are characterized by similar cyclization mechanisms as those observed for polyamine-derived alkaloids; they lead to the piperidines (six-ring) and quinolizidines (two fused six-rings) rather than the pyrrolidines (five-ring) and pyrrolizidines (two fused five-rings). A third class derived from lysine, the indolizidines, is characterized by a six-ring fused to a five-ring [
45]. Similar biosynthetic reaction mechanisms are also observed for the alkaloid classes derived from tyrosine and tryptophan. Decarboxylation of tyrosine and tryptophan leads to the phenylethylamines (e.g., hordenine) and the indolics (e.g., tryptamine). Schiff base formation with a short-chain aliphatic aldehyde/ketone followed by ring closure converts the phenylethylamines and the indolics to the tetrahydroisoquinolines and the β-carbolines, respectively. When Schiff base formation couples a phenylethylamine with a C
6C
2 aldehyde, such as phenylacetaldehyde, the benzyltetrahydroisoquinolines (e.g., reticuline) are produced, whereas coupling with a dihydro C
6C
3 aldehyde (e.g., 4-hydroxydihydrocinnamaldehyde) or with secologanin produces the phenethylisoquinolines and the terpenoid tetrahydroisoquinolines, respectively. Schiff base formation between indolics and secologanin yields the terpenoid indole alkaloids (e.g., strictosidine). Oxidative radical–radical coupling between the two phenolic moieties present in the structures of benzyltetrahydroisoquinolines and phenethylisoquinolines results in different skeleton types, i.e., the morphinan type (e.g., morphine) and the aporphine type, respectively. Another skeleton type, i.e., the protoberberine type (for example, berberine), is formed from benzyltetrahydroisoquinolines via a second Schiff base formation. Other alkaloid classes that are derived from tryptophan are the quinolines, pyrroloquinolines, pyrroloindoles and the ergot alkaloids [
45].
Figure 3. Biosynthesis of Alkaloids.
Within plants, alkaloids are believed to serve as a storage form of nitrogen and as protection against herbivory [
47]. When a pathogen or predator attacks a plant, the alkaloid can interfere with the predator’s protein synthesis, with the predator’s enzyme activity and/or with the predator’s nervous system [
48]. In addition, by inhibiting the growth of competing plants, alkaloids can act as an herbicide [
49]. The bioactivity of the alkaloids is due to the presence of the nitrogen atom; the lone pair electrons on the nitrogen can act as proton-acceptors, whereas the hydrogen-atoms in primary and secondary amines can act as a proton-donor. These properties lead to significant improvements in survival rates for plants [
48] and, at the same time, provide valuable biological properties for human therapeutics [
50]. Plant alkaloids are therefore widely known for their applications as drugs. For example, morphine, an alkaloid originally isolated from
Papaver somniferum (commonly known as opium poppy), is generally known for the treatment of severe pain. A dominant alkaloid class in monocots, such as maize, are the benzoxazinoids [
6,
51]. These indole-derived metabolites have been shown to be produced in response to European corn borer (
Ostrinia nubilalis)-herbivory [
52], and inoculation of maize leaves with pathogenic
Curvularia lunata resulted in the accumulation of HDMBOA-glucoside [
53].
1.4. Phenolics
Embryophytes, or land plants, appeared in the mid-Paleozoic era, between 480 and 360 million years ago. The successful adaptation of
Charophyceae from living underneath the water to living on land was achieved by the production of phenolic compounds, which act as antioxidants in the protection against UV radiation [
54]. The classification of phenolics is mainly based on their aromatic skeleton, e.g., C
6C
3 (phenylpropanoids), C
6C
3C
6 (flavonoids) or (C
6C
3C
6)
2 (proanthocyanidins, also called condensed tannins) [
54]. There are two biosynthetic pathways leading towards phenolic compounds, namely, via polyketide biosynthesis which mainly occurs in micro-organisms (
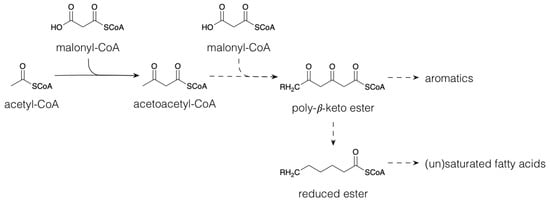
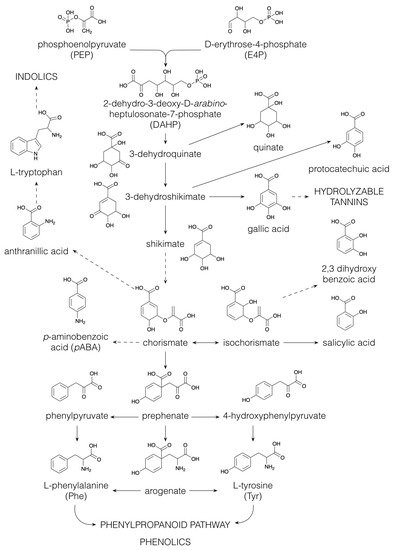
Figure 1), and via the shikimate pathway (
Figure 4), yielding the aromatic amino acids L-phenylalanine (Phe), L-tyrosine (Tyr) and L-tryptophan (Trp) [
55]. The shikimate pathway is employed by both micro-organisms and plants but does not exist in humans; therefore, the aromatic amino acids Phe, Tyr and Trp are essential for humans [
56]. The shikimate pathway (
Figure 4) starts with the aldol condensation of the glycolytic intermediate phosphoenolpyruvate and erythrose-4-phosphate, which is produced via either photosynthesis or the pentose phosphate pathway, to form 2-dehydro-3-deoxy-arabino-heptulosonate-7-phosphate (DAHP). Next, DAHP is converted to 3-dehydroquinate, which can be reduced to quinate or dehydrated to 3-dehydroshikimate, the latter being the precursor of the benzenoids protocatechuate and gallate [
55]. Gallate is, in turn, the starter-molecule for the biosynthesis of hydrolyzable tannins [
57]. Via shikimate, 3-dehydroshikimate is converted to chorismate, from which some simple aniline derivates are produced using glutamine as an amino-donor, e.g.,
p-aminobenzoic acid (
pABA).
pABA is part of the folic acid (vitamin B
9) structure, an essential compound in DNA synthesis, methylation and cellular division [
58]. Biofortification of maize by boosting folate biosynthesis has been demonstrated as an attractive strategy to enhance the nutritional value of maize [
59,
60]. Another aniline derivate, anthranilic acid, is an intermediate in the biosynthetic pathway to the aromatic amino acid Trp, which serves as a precursor for many indolics, for example, indole-3-acetic acid (IAA), a major regulator of plant growth [
61]. Isomerization of chorismate to isochorismate is the crucial step in the biosynthesis of the benzenoids, 2,3-dihydroxybenzoate and salicylate (see below). Furthermore, chorismate is an intermediate in the pathway towards the two aromatic amino acids Phe and Tyr. The first step in the biosynthesis of Phe and Tyr involves the conversion of chorismate to prephenate (
Figure 4). Then, two alternative pathways are possible. In the first, the arogenate pathway, prephenate, is transaminated to arogenate, followed by a dehydration and decarboxylation to Phe or a dehydration and a decarboxylation to Tyr. In an alternative route, the opposite occurs: prephenate is first dehydrated/dehydrogenated and decarboxylated to phenylpyruvate and
p-hydroxyphenylpyruvate and then transaminated to produce Phe and Tyr [
55]. Most microorganisms utilize the phenylpyruvate/
p-hydroxyphenylpyruvate pathways, whereas biosynthesis of Phe and Tyr in plants has been predominantly described to occur via the arogenate pathway [
62]. However, the alternative biosynthesis of Phe via phenylpyruvate, with Tyr as the amino-donor, has been described in
Petunia hybrida flowers [
63].
Figure 4. Biosynthesis of the aromatic amino acids via the shikimate pathway. Dashed arrows indicate multiple conversions.
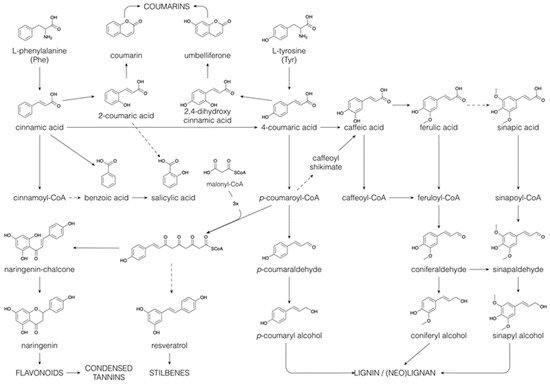
Phe and Tyr form the basis for the C
6C
3 phenylpropane unit that is found in a wide range of phenolic compounds, such as phenylpropanoids, flavonoids, coumarins, monolignols, lignins and lignans [
64]. The general phenylpropanoid pathway starts with the deamination of Phe leading to
trans-cinnamate (
Figure 5). Successive hydroxylations, by cytochrome P450 monooxygenases, and methylations, by methyltransferases, introduce hydroxyl and methoxyl substituents on the benzene ring, generating
p-coumaric acid (or 4-coumaric acid), caffeic acid, ferulic acid and sinapic acid [
55]. Following CoA esterification, the resulting phenylpropanoyl CoA esters can enter several of the above-mentioned specialized metabolic pathways, such as flavonoid and monolignol biosynthesis (see below). Besides their biosynthesis via the shikimate pathway, benzenoids can be produced from phenylpropanoids by shortening their side chains in a CoA-dependent β-oxidative pathway (analogous to the β-oxidation of fatty acids), or in a CoA-dependent or -independent non-oxidative pathway [
65]. The benzenoid salicylic acid can thus be synthesized (i) directly from chorismate, (ii) through isochorismate, (iii) by hydroxylation of benzoic acid (iv) or by side-chain cleavage of
o-coumaric acid (or 2-coumaric acid), which is formed by the 2-hydroxylation of cinnamic acid (
Figure 5, [
66]). Salicin, the precursor for the synthesis of acetylsalicylic acid, a widely used pain killer, is produced via reduction in the acidic function of salicylic acid, yielding salicylaldehyde, followed by glycosylation and a subsequent reduction in the aldehyde function to an alcohol [
67]. Salicin is the building block of the phenolic glycosides, a major compound class in the Salicaceae [
68].
Figure 5. Biosynthesis of coumarins, benzenoids, flavonoids, stilbenoids, lignins and (neo)lignans via the phenylpropanoid pathway. Dashed arrows indicate multiple conversions.
The ortho-hydroxylation of phenylpropanoyl CoA esters is essential in the formation of coumarins. The first step involves a cis–trans isomerization of the side-chain, which is then followed by lactonization [
55,
69]. As such, cinnamic acid and
p-coumaric acid give rise to coumarin and umbelliferone, respectively (
Figure 5). Further hydroxylation and methylation of the benzene ring yield esculetin and scopoletin, in which the position of the benzene substitutions reflects those found in caffeic acid and ferulic acid.
Reduction of the various phenylpropanoyl CoA esters to their corresponding aldehydes and further to their alcohols constitutes the monolignol biosynthetic pathway. The above-mentioned benzene ring-substitutions may also (partially) occur at the CoA, aldehyde or alcohol level rather than at the acid level [
69,
70]. Monolignols are exported to the cell wall, where they are oxidized by peroxidases and/or laccases to relatively stable, electron-delocalized radicals, and such monolignol radicals are also thought to be formed by oxidation within the cytoplasm [
4,
71]. Subsequent radical–radical coupling between monolignols, most often guided in a stereospecific way by a dirigent protein, leads to the synthesis of (neo)lignans. Coupling products derived from two monolignols that are linked via an 8–8 bond are referred to as lignans. If two monolignols are coupled via a different carbon–carbon bond, they are referred to as neolignans, and if they are linked via an ether linkage, they are referred to as oxyneolignans. Sesquineolignans are coupling products between three monolignols, and dineolignans are coupling products involving four monolignols [
72]. This structural variety of (neo)lignans is further increased by additional cyclizations, oxydations, hydroxylations and other modifications, resulting in a plethora of structures. Certain lignans, such as podophyllotoxin and its derivatives, have pronounced cytotoxic activity and are used as antitumor compounds [
73]. Although the biosynthesis of therapeutic lignans has been studied in depth, the role of these compounds in plants remains obscure today. Some, such as secoisolariciresinol, can serve as antioxidants; however, many others, such as justicidin, are the product of many oxidative reactions and so no antioxidant activity remains [
74]. Undirected, non-stereospecific radical–radical coupling between monolignols in the cell wall leads also to the formation of the lignin polymer [
75]. Depending on the plant species, the monolignols
p-coumaryl alcohol (yielding the H-unit in lignin), coniferyl alcohol (G-unit) and sinapyl alcohol (S-unit), and other minor monomers belonging to different phenolic classes (hydroxyarylpropanols, hydroxycinnamyl esters, hydroxycinnamaldehydes, hydroxycinnamic acids, hydroxycinnamate esters, hydroxycinnamides, hydroxybenzaldehydes, hydroxybenzoic acids, hydroxystilbenes and the flavone, tricin), can be incorporated in the lignin polymer, leading to a high, naturally occurring structural variation [
69,
70]. As lignins from more species and tissues continue to be examined, this structural variability is likely to continue growing. The flexibility of the lignin structure is further shown in mutants and transgenic plants. Upon monolignol biosynthesis pathway perturbation, intermediates accumulate and are converted into a wide range of derivatives, which are partially incorporated into the lignin polymer [
7,
69,
76,
77]. Lignin provides the plant with strength to grow upright, and with hydrophobicity for water transport, distributing nutrients from the soil within the plant. Although the incorporated monomers will affect the structure and physico-chemical properties of lignin, a large compositional variation in lignin is allowed without affecting its functionality [
69,
70,
78].
The phenylpropanoyl CoA esters can also serve as starter units for chain extension with malonyl CoA units. Chain extension involving three malonyl CoA molecules occurs most commonly and produces stilbenes (e.g., resveratrol) and chalcones (e.g., naringenin chalcone). Isomerization of the chalcones yields the flavanones (e.g., naringenin). This occurs by a Michael-type nucleophilic attack, yielding a six-membered heterocyclic ring. The flavanones can be converted to a wide range of flavonoids, i.e., flavones, flavonols, flavanols, proanthocyanidins, anthocyanins and catechins, among others, through hydroxylation, reduction and dehydration reactions of the heterocyclic ring. Flavonoids contribute to plant colours necessary for the attraction of pollinators [
79]. Chalcones and flavonols account for the yellow colors, whereas anthocyanidins are responsible for red and blue colors and combinations thereof. In addition, flavonoids are involved in plant development regulation, UV protection, plant-microorganism signaling and plant defense [
80].
A large portion of the structural diversity among polyphenolics results from oxidative coupling reactions, triggered by initial radical scavenging reactions. Combinatorial coupling between a flavonoid semiquinone radical and a phenylpropanoid radical, such as coniferyl alcohol, forms a flavonolignan [
111,
112]. Combinatorial radical–radical coupling among flavones, flavonols, dihydroflavonols, flavanones, isoflavones, aurones and chalcones forms bi-, tri-, and tetraflavonoids [
113,
114,
115,
116,
117,
118,
119,
120,
121]. Because the radical can be delocalized along the flavonoid conjugated system, various C–C or C–O–C linkage types are produced. These flavonoid oligomers can be distinguished from proanthocyanidin oligomers by the coupling mechanisms (proanthocyanidins are coupled through nucleophilic attacks) and by their monomers (proanthocyanidins are coupling products of flavan-3-ol monomers); however, the distinction is somehow arbitrary because “mixed” dimers, for example, flavan-3-ol → dihydroflavonol, and “nonproanthocyanidins” comprising oxidatively coupled flavan-3-ols have also been identified [
113]. Similarly to proanthocyanidins, dimer or oligomer molecules may exhibit enhanced activity relative to the simple monomer units. For example, bikaempferol showed stronger ROS scavenging activity than kaempferol [
121]. This amplifying effect can be induced by the increased number of phenolic hydroxyl groups and by the extension of the conjugated system over the two molecules. Not all plant extracted biflavonoids, however, exhibit strong antioxidant activity [
120]. In certain cases, dimerization may have led to new functionalities. Biflavonoids have been mainly studied for their strong cytotoxic activity, suggesting a defensive role against herbivores in plants [
119,
122,
123]. It is not known if the coupling occurs in a directed way, guided by a dirigent protein, in analogy with lignan biosynthesis, and also demonstrated for the synthesis of the phenolic terpenoid (+)-gossypol in cotton (
Gossypium sp.) [
124,
125]. In certain cases, however, racemic mixtures of biflavonoids have been reported, indicating an uncontrolled, non-stereospecific radical–radical coupling [
120]. Undirected radical–radical coupling in an oxidative cell environment could terminate radical chain reactions. The coupling products would then be, in some way, waste products with highly variable structures but, a priori, no proper function. In poplar, under oxidative conditions, monolignol radical coupling occurs within the cytoplasm [
4,
71]. Phenylcoumaran benzylic ether reductase (PCBER), an extremely abundant enzyme in poplar wood, reduces various products that arise from the oxidative coupling of monolignols, regenerating, as such, radical scavengers in an oxidative intracellular environment [
71]. Similar recycling loops of other types of polyphenolic radical coupling products could be highly efficient contributors of radical scavengers in plant cells, although confirmation of this functionality awaits, today, the functional characterization of more PCBER-like reductases in the plant specialized metabolism.
2. Decoration and Conjugation in Specialized Metabolism
2.1. Diversity of Decorations
The majority of the structural diversity in the plant specialized metabolome arises from “decorative” biosynthetic steps, such as hydroxylations, methylations, glycosylations, acylations and prenylations [126]. The number of different modifications is rather limited and the nature of the decorations for different classes of specialized metabolites is often the same. For example, classes as far apart as flavonoids, indole alkaloids and apocarotenoids are all decorated with glucose and with malonylglucose [1,3]. In the specialized metabolite profile of a plant extract, the common decorations are reflected in an overrepresentation of certain mass differences corresponding to the difference between two core structures. For example, in the maize specialized metabolome, the mass difference of 108.021 Da, corresponding to the difference between a phenylpropanoid and a flavonoid core structure, is observed between p-coumaric acid and naringenin, but also between caffeoyl hexose and eriodictyol 7-O-hexoside (hydroxylation and addition of a hexose to both) or between sinapoyl hexose and methoxy-homoeriodictyol (two methoxylations of both), as well as between dihydroxyindole-3-acetic acid (caffeoyl) hexoside and dihydroxyindole-3-acetic acid (eriodictyol-O-) hexoside (hydroxylation and addition of dihydroxyindole-3-acetic acid and hexose to both) [6]. The majority of plant specialized metabolites are glycosylated and/or acylated, with acylations being aromatic (p-coumaric, caffeic, ferulic, sinapic, gallic or p-hydroxybenzoic acids) or aliphatic (malonic, acetic, malic, succinic or oxalic acids). Because of the multitude of different possible decoration sites (hydroxyl groups), and the multitude of possible combinations of decorations, the number of possible structures grows exponentially with each added decoration [127]. Moreover, N-acylation of alkaloids, such as anthranilic acid or polyamines, leads to the formation of a high variety of hydroxycinnamic acid amides [128]. Intramolecular cyclization through phenol-oxidative coupling between the phenolic rings of two p-coumaroyl substitutes in bis-(p-coumaroyl) polyamines is the biosynthetic mechanism leading to cyclic alkaloids such as lunarine (in Lunaria annua seeds) and aphelandrine (in Aphelandra sp. roots) [129,130]. The introduction of phenolic rings through aromatic acylation allows, thus, further structural diversification through oxidative coupling.
2.2. High Molecular Weight Conjugates
Glucosylation and acylation allow for the concatenation of several specialized metabolites, sometimes of different metabolic classes. In the maize specialized metabolome, phenylpropanoid glycosides are coupled to benzoic acids, flavonoids, phenylethanoids and indolics [6]. Hexosylated flavonoids, but also free hexoses or dihexoses, often carry multiple aromatic acylations, often of different natures [3,6,131]. Less frequently, the hexose moiety in poly-acylated aromatic conjugates is replaced by an alternative polyol, such as glucaric acid [2]. The feruloyl moieties, both in glucaric acid and hexose conjugates, are sometimes coupled by radical–radical coupling to mono- or oligolignols, allowing these coupling products to be stored as such in the leaf vacuole [2,4]. In contrast to glucaric acid conjugates, where the two carboxylic acid groups of glucaric acid remain unesterified, other aromatic conjugates involve esterification to the carboxyl groups of dicarboxylic acids such as succinic acid or 3-hydroxy-3-methylglutaric acid. For example, in blue Agapanthus flowers, a p-coumaroylated delphinidin diglycoside is attached to a favonol triglycoside via a succinic acid diester link [132]. In this conjugate, the aromatic acyl group and the colourless flavonoid both stabilize the blue color of the anthocyanin delphidin diglycoside. Esterification involving the dicarboxylic acids 3-hydroxy-3-methylglutaric acid (HMGA) and succinic acid allowed the formation of a macromolecular polyester structure carrying glucosylated lignans, glucosylated flavonoids, phenylpropanoic acids and ferulic acid coupled oligolignols in the outer integument of flax seeds [127]. More common polyesters in higher plants are cutin and suberin, of which the latter contains aromatic domains derived from cinnamic acids [133]. Cutin and suberin function as protecting barriers against desiccation or biotic and abiotic stresses.