1. Cisplatin as a Treatment Modality for Cancer
The use of cisplatin as an anticancer agent was first published in 1969, describing its action against malignant murine sarcoma and leukemia
[1]. Higby and Wallace investigated cisplatin in metastatic testicular cancer, wherein they reported seven cases of complete recovery and 13 cases of significant tumor regression in a 15-patient clinical study
[2]. Einhorn and Donohue combined cisplatin with bleomycin and vinblastine for advanced testicular cancer
[3]. This three-drug regimen had an initial 70% complete re-sponse rate and five-year survival rate of 64%
[3]. Wiltshaw and colleagues reported similar outcomes for advanced ovarian cancer using cisplatin as a single agent in 82 ovarian cancer patients previously treated with conventional chemotherapy
[4]. Ovarian cancer response rate was dose-dependent, ranging between 33% for a 30 mg/m
2 dose and 52% for a 100 mg/m
2 dose
[4]. On the basis of the success of these trials, cisplatin expanded to include additional malignancies such as cervical, lung, and head and neck cancers
[5]. The outcomes were consistent with the testicular and ovarian cancer studies, wherein cisplatin was effective both as a single agent and in combination with other chemotherapeutic agents
[6].
With cisplatin’s promising clinical trial success as an anti-cancer therapy, it became vital to understand this novel drug’s underlying mechanism of action. In 1970, Rosenberg and VanCamp proposed that cisplatin stimulated an immune response
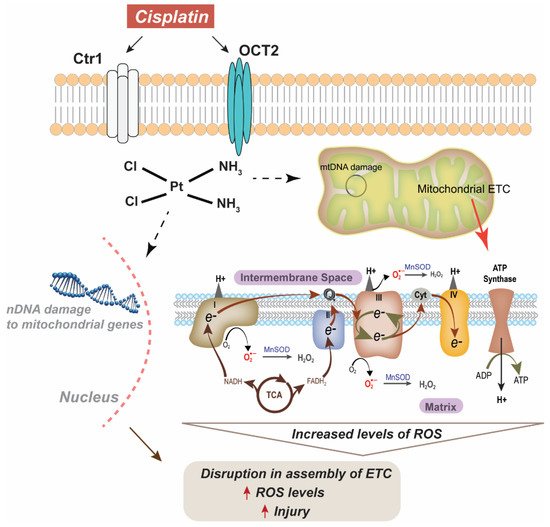
[7]. Later studies in mammalian cells and animals treated with cisplatin then revealed that the drug inhibits DNA synthesis and cell growth
[8][9]. This discovery was made by tracing the incorporation of the radioactive DNA, RNA, and protein precursors 3H-thymidine, 3H-uridine, and 3H-L-leucine, respectively. Cisplatin hindered 3H-thymidine incorporation into DNA but not 3H-uridine or 3H-L-leucine incorporation both in vitro and in vivo
[8][9]. It is now established that cisplatin binds to DNA purines at the N7 position and forms 1-, 2-, or 3-intrastrand crosslinks that terminate DNA replication and transcription and recruit high-mobility group box protein 1 (HMGB1), leading to the activation of pathways associated with DNA damage and apoptosis, such as p53 and MAPK
[10].
Another noteworthy aspect of cisplatin’s history as an anti-cancer therapy is its radiation sensitizing activity. In 1978, Alvarez and colleagues reported that cisplatin sensitized TC.SV-40 cells against ionizing radiation in vitro
[11]. As cisplatin showed efficacy as a chemotherapeutic agent in clinical trials, it was also tested in combination with radiotherapy. In 1981, 124 patients with advanced inoperable squamous cell carcinoma of the head and neck received cisplatin (100 mg/m
2) every three weeks concurrently with definitive radiotherapy (planned total dose ≥ 64.5 Gy)
[12]. Patients in this trial had significantly improved clinical response rates that differed on the basis of tumor site and differentiation state. Patients with hypopharyngeal cancer responded 25% of the time, while patients with nasopharyngeal tumors responded 83% of the time. The response rate for poorly differentiated tumors was 89% compared to 67% and 59% for well-differentiated and moderately differentiated tumors, respectively
[10]. However, severe toxicities associated with this treatment regimen included leukopenia (11%), nausea and vomiting (8%), stomatitis (31%), and nephrotoxicity (6%)
[12].
Subsequent randomized clinical trials have shown concurrent cisplatin improves locoregional control, progression-free survival, and overall survival in non-small cell lung cancer
[13], cervical cancer
[14], and head and neck cancer
[15][16][17] over radiation alone, induction chemotherapy, or radiation in combination with other agents. Rates of severe toxicities from concurrent cisplatin in these trials include leukopenia (11–42%), nausea and vomiting (8–28%), stomatitis (31–43%), anemia (17%), dermatitis (7%), neurologic toxicity (5%), and nephrotoxicity (4–8%)
[12][13][14][15][16][17]. Any grade acute kidney injury incidence is as high as 34% with high dose cisplatin (100 mg/m
2 q3 weeks)
[18]. The risk of cisplatin-induced nephrotoxicity increases with cisplatin dose and duration of treatment
[19]. For example, 34% of head and neck cancer patients treated with fractionated ionizing radiation (total dose of 60 to 70 Gy in 2 Gy fractions) and cisplatin therapy (100 mg/m
2 delivered every 21 days for 3 cycles) develop cisplatin-induced AKI
[18]. A decline in renal function may necessitate cisplatin administration delays and dose reductions as patients cannot receive a planned dose of cisplatin
[18]. Risk factors for developing nephrotoxicity following cisplatin exposure are related to the renal clearance of cisplatin. Patients prone to developing AKI following cisplatin treatment include those that have high peak plasma cisplatin concentrations (>400 ng/mL)
[16], pre-existing kidney damage (creatinine > 1.5 mg/dL)
[17], age ≥ 61 years, and a history of hypertension
[20][21]. Survivors of childhood cancers treated with cisplatin (cumulative doses > 450 mg) develop long-term (decades) nephrotoxicity with reduced estimated glomerular filtration rates compared to childhood cancer survivors not treated with cisplatin (eGFR of 83 mL/min/1.73 m
2 vs. 101 mL/min/1.73 m
2). Adult cancer survivors treated with cisplatin are also prone to worsening long term renal function and chronic kidney disease. A retrospective review of 777 adult cancer survivors treated with cisplatin had an average eGFR reduction of 0.73 mL/min per 1.73 m
2 per year.
RTOG-1016 randomized 849 subjects with locally advanced oropharyngeal carcinoma to receive radiotherapy (70 Gy/35 fx) combined with either cisplatin (100 mg/m
2 on days 1 and 22 of radiation) or cetuximab (loading dose of 400 mg/m
2 for 5–7 days followed by weekly cetuximab at 250 mg/m
2 for seven doses). Patients treated with cisplatin had an improved 5-year progression-free survival (78% vs. 67%) and reduced 5-year local regional failure (9.9% vs. 17%). There were no significant differences in xerostomia, fibrosis, muscle atrophy, and weight loss. On the basis of these data, the research found that radiation combined with cisplatin is superior to radiation combined with cetuximab for the definitive treatment of locally advanced oropharyngeal carcinoma
[16].
Despite its treatment efficacy, cisplatin treatment is known to cause significant toxicities. A phase III intergroup trial in head and neck cancer patients comparing subjects that received radiation alone (70 Gy/35 fx), radiation and cisplatin (100 mg/m
2 on days 1, 22, and 43), or split course radiation was given with three cycles of 5-fluorouracil and cisplatin chemotherapy, identifying improved 3-year overall survival in patients treated with concurrent cisplatin and radiation
[12]. Relative to subjects receiving radiation alone, however, subjects treated with concurrent cisplatin and radiation had an increased risk for ≥ grade 3 nausea and vomiting (16% vs. 6%), leukopenia (42% vs. 1%), anemia (17% vs. 0%), and nephrotoxicity (8% vs. 1%).
A retrospective review of 821 adult cancer survivors treated with cisplatin who survived for at least 5 years demonstrated the following changes in renal function: patients who were CKD stage 1 pre-cisplatin treatment progressed to CKD stage 2 (48%) or CKD stage 3 (14%), while only 36% remained at CKD stage 1
[22].
A common clinical approach to prevent and reduce the severity of cisplatin-associated nephrotoxicity is pre-hydration with intravenous isotonic saline to increase diuresis
[23]. Additional common clinical approaches include avoiding concomitant nephrotoxic drugs, reducing cisplatin dose
[24], and substituting an alternative chemotherapy agent for cisplatin
[25]. Examples of additional approaches that are less commonly utilized clinically include amifostine and theophyilline. Amifostine is approved by the FDA to reduce renal injury associated with multiple cisplatin administrations
[26][27]. Amifostine is a thiol derivative that scavenges free radicals generated during radiation and chemotherapy
[28]. Pre-clinical studies demonstrate that amifostine reduces mitochondrial membrane potential and reactive oxygen species formation in murine hepatocytes but not in hepatoma cells
[29]. However, because amifostine has a short half-life and significant side effects (nausea, vomiting, and hypotension), it is rarely used clinically
[26]. Theophylline is a competitive inhibitor of the adenosine receptor
[30]. Adenosine reduces GFR by constricting afferent arterioles, and preclinical studies demonstrated that adenosine receptor antagonists reduced acute renal injury
[31][32]. A randomized, single-blinded, placebo-controlled trial in 41 patients receiving cisplatin (50 mg/m
2) as part of their chemotherapy regimen demonstrated that theophylline preserved GFR compared to placebo-controlled subjects
[30].
Whether as a single chemotherapeutic agent, in combination with other chemotherapies, or in combination with ionizing radiation, cisplatin is still considered one of the most essential and reliable treatment agents for numerous malignancies. However, cisplatin-associated toxicities, especially nephrotoxicity, can dramatically hinder individual patient clinical outcomes; therefore, research dedicated to understanding and overcoming cisplatin toxicity is critical.
2. Characterization of Kidney Injury
The Kidney Disease: Improving Global Outcomes (KDIGO) guidelines define AKI as an abrupt decrease in kidney function that occurs over a period of 7 days or less, and CKD as abnormalities in kidney structure or function that persist for >90 days
[33][34]. Acute kidney disease (AKD) is described by KDIGO as acute or subacute damage or loss of kidney function for a duration of between 7 and 90 days after exposure to an AKI-initiating event
[33][34]. Several definitions of AKI have been validated, including the risk, injury, failure, loss of kidney function, and end-stage kidney disease (RIFLE) classification based on serum creatinine (sCr) or urinary outputs (UO) (
Table 1)
[35], with the acute kidney in-jury network (AKIN) classification being based on a ≥50% increase in absolute sCr (1.5 × baseline value) or a decrease in UO to <0.5 mL/kg/h for more than six hours
[35]. The AKIN classification uses the staging system described in
Table 1. After diagnosis of AKI by either classification, the KDIGO guidelines suggest monitoring sCr and UO for three months for resolution, new-onset, or worsening kidney dysfunction leading to chronic kidney disease (CKD)
[18]. Criteria to meet the definition of CKD is determined by duration; glomerular filtration rate (GFR); and abnormal urinalysis, pathology, or structure of the kidneys
[36]. CKD staging is based on GFR (mL/min/1.73 m
2) and the presence of albuminuria (
Table 2). Hypertension, diabetes, and hypercholesterolemia are risk factors for the development of CKD. Current CKD staging is based on GFR (mL/min/1.73 m
2) and presence of albuminuria (
Table 2). Long-term kidney dysfunction is notable in 60–80% of patients who receive cisplatin chemotherapy
[37].
Table 1. AKIN vs. RIFLE classification for kidney injury based on serum creatinine (sCr) and/or urinary outputs (UO).
| AKIN |
UO (Common to Both) |
RIFLE |
| Stage 1 Increase of ≥ 0.3 mg/dl or increase in more than or equal to 150–200% from baseline. |
Less than 0.5 mg/kg/L per hour for more than 6 h |
Risk Increase in sCr × 1.5 or GFR decrease >25% |
| Stage 2 Increase to more than 200–300% from baseline. |
Less than 0.5 mg/kg/L per hour for more than 12 h |
Injury sCr × 2 or GFR decrease >50% |
| Stage 3 Increased to more than 300% from baseline with an acute increase of at least 0.5 mg/dL or on RRT. |
Less than 0.3 mg/kg/L for 24 h or anuria for 12 h |
Failure sCr × 3 or >4 mg/dL with an acute rise >0.5 mg/dL or GFR decrease >75% |
| |
|
Loss Persistent acute kidney failure = complete loss of kidney function >4 weeks |
| |
|
End-Stage Kidney Disease ESKD >3 months |
AKIN, Acute Kidney Injury Network; ESKD, end-stage kidney disease; GFR, glomerular filtration rate; sCr, serum creatinine; RIFLE, risk, injury, failure, loss, and end stage; RRT, renal replacement therapy.
Table 2. Staging system for chronic kidney disease as per Kidney Disease Improving Global Outcomes (KDIGO) guidelines.
| GFR Stages |
Kidney Function |
GFR (mL/min/1.73 m2) |
| Stage G1 |
Normal |
≥90 |
| Stage G2 |
Mildly Decreased |
60–90 |
| Stage G3a |
Mildly to Moderately Decreased |
45–59 |
| Stage G3b |
Moderately to Severely Decreased |
30–44 |
| Stage G4 |
Severely Decreased |
15–29 |
| Stage G5 |
Kidney Failure |
<15 |
3. Pathophysiology of AKI and CKD
While the term “AKI” is clinical, the use of acute tubular injury (ATI) is used to classify kidney injury histopathologically. In practice, ATI is semi-quantified as either mild, moderate, or severe injury and as well as focal vs. diffuse injury
[38]. Characterization of kidney biopsy samples for ATI is made by the presence of tubular luminal dilation, loss of the brush border in tubules, loss of nuclei, and the presence of cytoplasmic basophilia
[38]. Additionally, distinct pathological markers can be found in AKI associated with pigment administration, crystallopathy, nephrotoxic drug administration, and infection
[38]. An increase in pathophysiology studies has revealed that oxidative stress, endothelial injury, mitochondrial injury, and immunological responses are key mechanisms to the AKI development of AKI. Furthermore, AKI is now considered a prominent risk factor for the CKD development of CKD, particularly in older patients and patients who have had multiple AKI episodes
[38].
The definition of CKD includes not only decreases in GFR, but also structural and functional abnormalities of the kidney. Functional abnormalities such as albuminuria, proteinuria, and hematuria are classic examples. Glomerular filtration is highly dependent on high intra- and trans-glomerular pressure, which is reflected in hemodynamic injury to the kidney
[39]. Additionally, CKD is promoted when the glomerular membrane’s electrostatic barrier is disrupted, allowing proteins to move into Bowman’s capsule
[39]. Tubulointerstitial impairment also closely associates with long-term kidney dysfunction and encompasses many pathological features such as interstitial inflammation, kidney fibrogenesis, fibroblast activation, and promotion of the epithelial–mesenchymal transition (EMT)
[39].
 +1 credit
+1 credit