 +1 credit
+1 credit
Video Upload Options
Megalin, a recently discovered endocytic receptor for prorenin and renin, not only contributes to renin/prorenin reabsorption in the proximal tubule of the kidney, but simultaneously plays a role in renal Ang II generation, although how exactly this occurs is still unknown .
1. Introduction
2. Expression of RAS Components in Healthy and Pre-Eclamptic Placentas
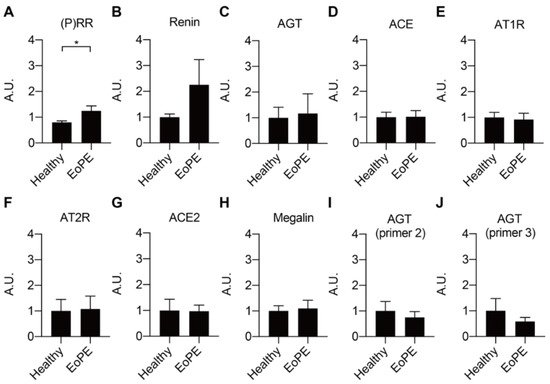
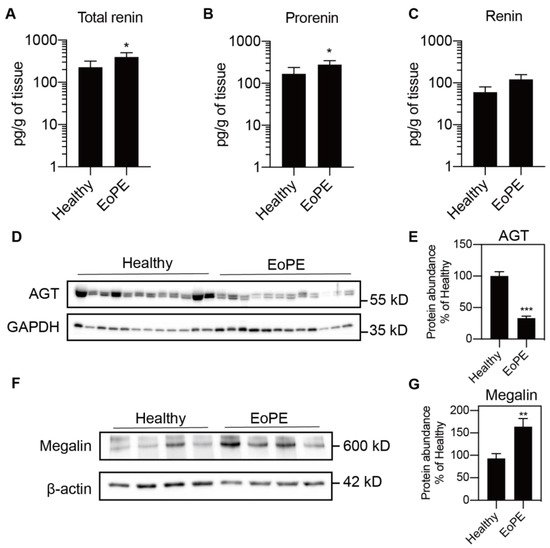
Placental expression of renin, AGT, angiotensin-converting enzyme (ACE), ACE2, the Ang II type 1 and type 2 receptor (AT1R, AT2R), and megalin was comparable between healthy and pre-eclamptic placentas (n = 12 for each), while (P)RR expression was increased in the latter (Figure 1A–H). AGT expression was at or below the detection limit (Ct = 38) in most of the samples, even when using two additional sets of primers (Figure 1I,J). Total renin levels in healthy placental tissue amounted to 118 (range 42.5–1476) pg/g tissue, and 72 ± 2% of this was prorenin (Figure 2A–C; n = 16). Total renin levels were significantly (p < 0.05) higher in pre-eclamptic placentas (266 (range 54.5–1568) pg/g tissue), and this was due to upregulated prorenin (p < 0.05) rather than renin. Nevertheless, the proportion of renin in the pre-eclamptic group (29 ± 2%) was not different from that in healthy placentas. Placental AGT abundance was decreased (p < 0.0001) in pre-eclamptic placentas (Figure 2D,E), while the opposite was true (p < 0.01) for megalin protein abundance (Figure 2F,G).
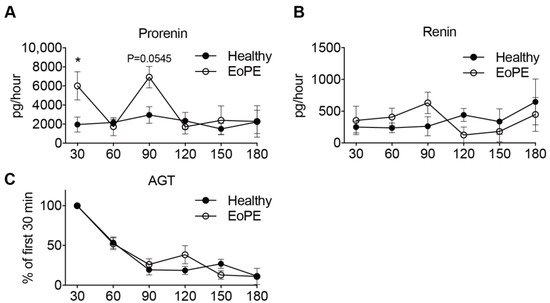
3. Placental Release of (Pro)renin and AGT
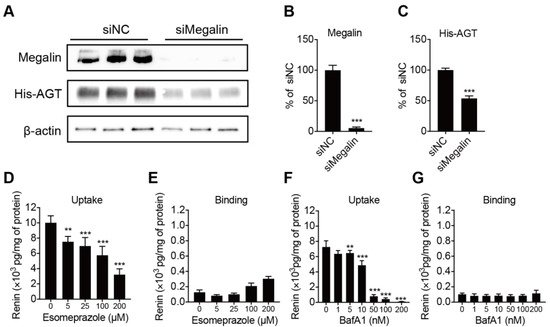
4. Megalin Internalizes AGT and PPI Decreases Renin Internalization but Not Binding in BN16 Cells
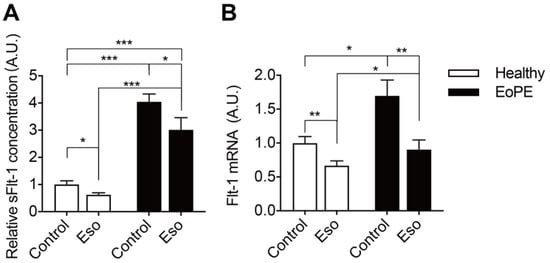
5. PPI Reduces sFlt-1 Secretion in Human Placental Villous Explants
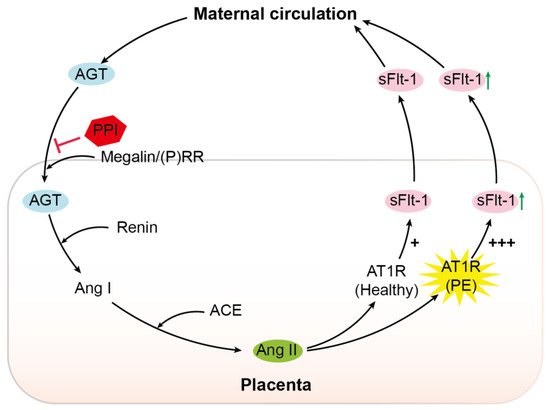
6. Discussion
References
- Mol, B.W.J.; Roberts, C.T.; Thangaratinam, S.; Magee, L.A.; de Groot, C.J.M.; Hofmeyr, G.J. Pre-eclampsia. Lancet 2016, 387, 999–1011.
- Miklus, R.M.; Elliott, C.; Snow, N. Surgical cricothyrotomy in the field: Experience of a helicopter transport team. J. Trauma 1989, 29, 506–508.
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289.
- Verdonk, K.; Visser, W.; Van Den Meiracker, A.H.; Danser, A.H. The renin-angiotensin-aldosterone system in pre-eclampsia: The delicate balance between good and bad. Clin. Sci. 2014, 126, 537–544.
- Rana, S.; Powe, C.E.; Salahuddin, S.; Verlohren, S.; Perschel, F.H.; Levine, R.J.; Lim, K.H.; Wenger, J.B.; Thadhani, R.; Karumanchi, S.A. Angiogenic factors and the risk of adverse outcomes in women with suspected preeclampsia. Circulation 2012, 125, 911–919.
- Derkx, F.H.; Alberda, A.T.; de Jong, F.H.; Zeilmaker, F.H.; Makovitz, J.W.; Schalekamp, M.A. Source of plasma prorenin in early and late pregnancy: Observations in a patient with primary ovarian failure. J. Clin. Endocrinol. Metab. 1987, 65, 349–354.
- Hsueh, W.A.; Luetscher, J.A.; Carlson, E.J.; Grislis, G.; Fraze, E.; McHargue, A. Changes in active and inactive renin throughout pregnancy. J. Clin. Endocrinol. Metab. 1982, 54, 1010–1016.
- Irani, R.A.; Xia, Y. Renin angiotensin signaling in normal pregnancy and preeclampsia. Semin. Nephrol. 2011, 31, 47–58.
- Sealey, J.E.; McCord, D.; Taufield, P.A.; Ales, K.A.; Druzin, M.L.; Atlas, S.A.; Laragh, J.H. Plasma prorenin in first-trimester pregnancy: Relationship to changes in human chorionic gonadotropin. Am. J. Obstet. Gynecol. 1985, 153, 514–519.
- Krop, M.; Danser, A.H. Circulating versus tissue renin-angiotensin system: On the origin of (pro)renin. Curr. Hypertens. Rep. 2008, 10, 112–118.
- Irani, R.A.; Xia, Y. The functional role of the renin-angiotensin system in pregnancy and preeclampsia. Placenta 2008, 29, 763–771.
- Verdonk, K.; Saleh, L.; Lankhorst, S.; Smilde, J.E.; van Ingen, M.M.; Garrelds, I.M.; Friesema, E.C.; Russcher, H.; van den Meiracker, A.H.; Visser, W.; et al. Association studies suggest a key role for endothelin-1 in the pathogenesis of preeclampsia and the accompanying renin-angiotensin-aldosterone system suppression. Hypertension 2015, 65, 1316–1323.
- Brar, H.S.; Kjos, S.L.; Dougherty, W.; Do, Y.S.; Tam, H.B.; Hsueh, W.A. Increased fetoplacental active renin production in pregnancy-induced hypertension. Am. J. Obstet. Gynecol. 1987, 157, 363–367.
- Kalenga, M.K.; Thomas, K.; de Gasparo, M.; De Hertogh, R. Determination of renin, angiotensin converting enzyme and angiotensin II levels in human placenta, chorion and amnion from women with pregnancy induced hypertension. Clin. Endocrinol. 1996, 44, 429–433.
- Herse, F.; Dechend, R.; Harsem, N.K.; Wallukat, G.; Janke, J.; Qadri, F.; Hering, L.; Muller, D.N.; Luft, F.C.; Staff, A.C. Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension 2007, 49, 604–611.
- Pringle, K.G.; Wang, Y.; Lumbers, E.R. The synthesis, secretion and uptake of prorenin in human amnion. Physiol. Rep. 2015, 3.
- Koizumi, M.; Ueda, K.; Niimura, F.; Nishiyama, A.; Yanagita, M.; Saito, A.; Pastan, I.; Fujita, T.; Fukagawa, M.; Matsusaka, T. Podocyte Injury Augments Intrarenal Angiotensin II Generation and Sodium Retention in a Megalin-Dependent Manner. Hypertension 2019, 74, 509–517.
- Storm, T.; Christensen, E.I.; Christensen, J.N.; Kjaergaard, T.; Uldbjerg, N.; Larsen, A.; Honore, B.; Madsen, M. Megalin Is Predominantly Observed in Vesicular Structures in First and Third Trimester Cytotrophoblasts of the Human Placenta. J. Histochem. Cytochem. 2016, 64, 769–784.
- Gleixner, E.M.; Canaud, G.; Hermle, T.; Guida, M.C.; Kretz, O.; Helmstadter, M.; Huber, T.B.; Eimer, S.; Terzi, F.; Simons, M. V-ATPase/mTOR signaling regulates megalin-mediated apical endocytosis. Cell Rep. 2014, 8, 10–19.
- Hastie, R.; Bergman, L.; Cluver, C.A.; Wikman, A.; Hannan, N.J.; Walker, S.P.; Wikstrom, A.K.; Tong, S.; Hesselman, S. Proton Pump Inhibitors and Preeclampsia Risk Among 157,720 Women. Hypertension 2019, 73, 1097–1103.
- Saleh, L.; Samantar, R.; Garrelds, I.M.; van den Meiracker, A.H.; Visser, W.; Danser, A.H.J. Low Soluble Fms-Like Tyrosine Kinase-1, Endoglin, and Endothelin-1 Levels in Women With Confirmed or Suspected Preeclampsia Using Proton Pump Inhibitors. Hypertension 2017, 70, 594–600.
- Zhou, C.C.; Ahmad, S.; Mi, T.; Xia, L.; Abbasi, S.; Hewett, P.W.; Sun, C.; Ahmed, A.; Kellems, R.E.; Xia, Y. Angiotensin II induces soluble fms-Like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancy. Circ. Res. 2007, 100, 88–95.
- Spugnini, E.P.; Citro, G.; Fais, S. Proton pump inhibitors as anti vacuolar-ATPases drugs: A novel anticancer strategy. J. Exp. Clin. Cancer Res. 2010, 29, 44.
- Sabolic, I.; Brown, D.; Verbavatz, J.M.; Kleinman, J. H(+)-ATPases of renal cortical and medullary endosomes are differentially sensitive to Sch-28080 and omeprazole. Am. J. Physiol. 1994, 266, F868–F877.
- Onda, K.; Tong, S.; Beard, S.; Binder, N.; Muto, M.; Senadheera, S.N.; Parry, L.; Dilworth, M.; Renshall, L.; Brownfoot, F.; et al. Proton Pump Inhibitors Decrease Soluble fms-Like Tyrosine Kinase-1 and Soluble Endoglin Secretion, Decrease Hypertension, and Rescue Endothelial Dysfunction. Hypertension 2017, 69, 457–468.
- Sun, Y.; Goes Martini, A.; Janssen, M.J.; Garrelds, I.M.; Masereeuw, R.; Lu, X.; Danser, A.H.J. Megalin: A Novel Endocytic Receptor for Prorenin and Renin. Hypertension 2020, 75, 1242–1250.
- Poisner, A.M. Regulation of utero-placental prorenin. Adv. Exp. Med. Biol. 1995, 377, 411–426.
- Shah, D.M.; Banu, J.M.; Chirgwin, J.M.; Tekmal, R.R. Reproductive tissue renin gene expression in preeclampsia. Hypertens. Pregnancy 2000, 19, 341–351.
- Anton, L.; Brosnihan, K.B. Systemic and uteroplacental renin--angiotensin system in normal and pre-eclamptic pregnancies. Ther. Adv. Cardiovasc. Dis. 2008, 2, 349–362.
- Anton, L.; Merrill, D.C.; Neves, L.A.; Stovall, K.; Gallagher, P.E.; Diz, D.I.; Moorefield, C.; Gruver, C.; Ferrario, C.M.; Brosnihan, K.B. Activation of local chorionic villi angiotensin II levels but not angiotensin (1-7) in preeclampsia. Hypertension 2008, 51, 1066–1072.
- Anton, L.; Merrill, D.C.; Neves, L.A.; Diz, D.I.; Corthorn, J.; Valdes, G.; Stovall, K.; Gallagher, P.E.; Moorefield, C.; Gruver, C.; et al. The uterine placental bed Renin-Angiotensin system in normal and preeclamptic pregnancy. Endocrinology 2009, 150, 4316–4325.
- Li, C.; Ansari, R.; Yu, Z.; Shah, D. Definitive molecular evidence of renin-angiotensin system in human uterine decidual cells. Hypertension 2000, 36, 159–164.
- Nonn, O.; Fischer, C.; Geisberger, S.; El-Heliebi, A.; Kroneis, T.; Forstner, D.; Desoye, G.; Staff, A.C.; Sugulle, M.; Dechend, R.; et al. Maternal Angiotensin Increases Placental Leptin in Early Gestation via an Alternative Renin-Angiotensin System Pathway: Suggesting a Link to Preeclampsia. Hypertension 2021, 77, 1723–1736.
- Takimoto, E.; Ishida, J.; Sugiyama, F.; Horiguchi, H.; Murakami, K.; Fukamizu, A. Hypertension induced in pregnant mice by placental renin and maternal angiotensinogen. Science 1996, 274, 995–998.
- Haase, N.; Foster, D.J.; Cunningham, M.W.; Bercher, J.; Nguyen, T.; Shulga-Morskaya, S.; Milstein, S.; Shaikh, S.; Rollins, J.; Golic, M.; et al. RNA interference therapeutics targeting angiotensinogen ameliorate preeclamptic phenotype in rodent models. J. Clin. Invest. 2020, 130, 2928–2942.
- Lenz, T.; James, G.D.; Laragh, J.H.; Sealey, J.E. Prorenin secretion from human placenta perfused in vitro. Am. J. Physiol. 1991, 260, E876–E882.
- Lenz, T. Release of prorenin and placental hormones from superfused minced chorion laeve. Acta Obstet. Gynecol. Scand. 1997, 76, 903–906.
- Ye, F.; Wang, Y.; Wu, C.; Howatt, D.A.; Wu, C.H.; Balakrishnan, A.; Mullick, A.E.; Graham, M.J.; Danser, A.H.J.; Wang, J.; et al. Angiotensinogen and Megalin Interactions Contribute to Atherosclerosis-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 150–155.
- Zygmunt, M.; McKinnon, T.; Herr, F.; Lala, P.K.; Han, V.K. HCG increases trophoblast migration in vitro via the insulin-like growth factor-II/mannose-6 phosphate receptor. Mol. Hum. Reprod. 2005, 11, 261–267.
- Burke, S.D.; Zsengeller, Z.K.; Khankin, E.V.; Lo, A.S.; Rajakumar, A.; DuPont, J.J.; McCurley, A.; Moss, M.E.; Zhang, D.; Clark, C.D.; et al. Soluble fms-like tyrosine kinase 1 promotes angiotensin II sensitivity in preeclampsia. J. Clin. Invest. 2016, 126, 2561–2574.


