Pre-eclampsia is a hypertensive disorder characterized by new-onset hypertension and proteinuria occurring after 20 weeks of gestation. It is one of the major causes of worldwide maternal mortality and morbidity [
1,
2,
3]. A normal pregnancy requires a ≈30% increase in extracellular fluid volume to provide an adequate blood supply for the developing uterus, placenta and fetus [
4]. The renin–angiotensin system (RAS) plays a key role in this process. Estrogen stimulates hepatic angiotensinogen (AGT) expression, resulting in a 3–5-fold rise in circulating AGT [
4,
5]. The plasma renin concentration also rises modestly [
6], and together with the rise in AGT, this results in a significant rise in plasma renin activity (PRA); thus, ensuring sufficient RAS activity to allow water and salt retention [
7,
8]. Remarkably, plasma prorenin levels increase much more strongly than plasma renin levels [
6,
9], due to prorenin release from the ovaries and, to a lesser degree, the placenta [
10]. The function of this prorenin remains unclear.
In pre-eclampsia, the rise in renin and AGT in the circulation is suppressed, although sensitivity to angiotensin (Ang) II is enhanced [
11]. Circulating prorenin levels in pre-eclamptic women are in the normal pregnancy range [
12]. Given the local production of prorenin in the placenta, while the placenta is a key player in the development of pre-eclampsia, disturbed placental RAS activity may contribute to this phenomenon. Yet, data on prorenin levels in the uteroplacental unit of pre-eclamptic women are conflicting, with evidence for decreases, increases and no alteration [
13,
14,
15]. What exactly determines the distribution of prorenin across the ovaries and uteroplacental unit, as well as its release into the blood stream, is unknown. It may require transcellular transport. For instance, amniotic fluid prorenin is of chorionic origin [
16], implying that chorionic prorenin is capable of crossing the amnion membrane. A novel player in this field is megalin. Megalin, a recently discovered endocytic receptor for prorenin and renin, not only contributes to renin/prorenin reabsorption in the proximal tubule of the kidney, but simultaneously plays a role in renal Ang II generation, although how exactly this occurs is still unknown [
17]. Outside the kidney, megalin is present in syncytiotrophoblast and cytotrophoblast cells in the placenta [
18]. Membrane recycling of megalin is a fast process which relies on vacuolar H
+-ATPase (V-ATPase)-dependent endosomal acidification [
19]. The latter involves the so-called (pro)renin receptor ((P)RR), which is an accessory protein of V-ATPase. Here, it is of interest to note that proton pump inhibitors (PPIs) have been observed to lower the placental release of soluble Fms-like tyrosine kinase-1 (sFlt-1), a mediator of maternal endothelial dysfunction in pre-eclampsia [
20,
21]. Ang II is a stimulator of sFlt-1 [
22]. Although PPIs are capable of inhibiting V-ATPase [
23,
24], such inhibition did not contribute to their acute effects on sFlt-1 release from human trophoblast cells [
25]. A further possibility is that PPIs affect sFlt-1 production indirectly, by preventing RAS component transport/uptake.
2. Expression of RAS Components in Healthy and Pre-Eclamptic Placentas
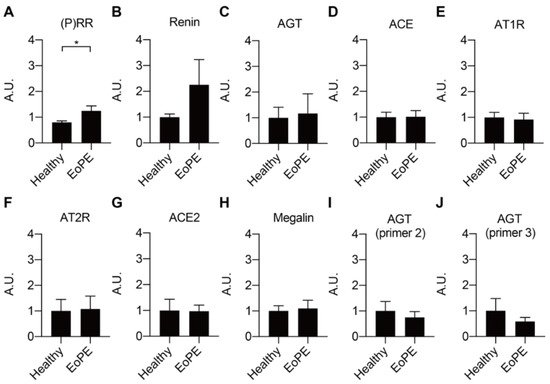
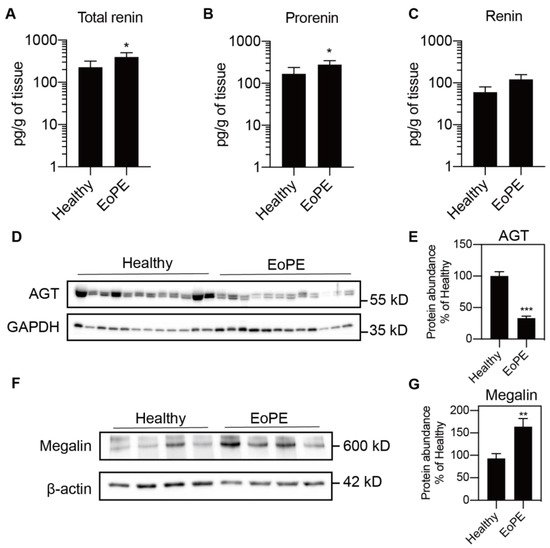
Placental expression of renin, AGT, angiotensin-converting enzyme (ACE), ACE2, the Ang II type 1 and type 2 receptor (AT1R, AT2R), and megalin was comparable between healthy and pre-eclamptic placentas (n = 12 for each), while (P)RR expression was increased in the latter (Figure 1A–H). AGT expression was at or below the detection limit (Ct = 38) in most of the samples, even when using two additional sets of primers (Figure 1I,J). Total renin levels in healthy placental tissue amounted to 118 (range 42.5–1476) pg/g tissue, and 72 ± 2% of this was prorenin (Figure 2A–C; n = 16). Total renin levels were significantly (p < 0.05) higher in pre-eclamptic placentas (266 (range 54.5–1568) pg/g tissue), and this was due to upregulated prorenin (p < 0.05) rather than renin. Nevertheless, the proportion of renin in the pre-eclamptic group (29 ± 2%) was not different from that in healthy placentas. Placental AGT abundance was decreased (p < 0.0001) in pre-eclamptic placentas (Figure 2D,E), while the opposite was true (p < 0.01) for megalin protein abundance (Figure 2F,G).
Figure 1. Gene expression levels of RAS components (A–G) and megalin (H) in healthy and early-onset pre-eclamptic (EoPE) placental tissue. (I,J): AGT expression was confirmed with different pairs of primers. Data are mean ± SEM of n = 12. * p < 0.05. (P)RR, (pro)renin receptor; AGT, angiotensinogen; ACE, angiotensin-converting enzyme; AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; A.U., arbitrary unit.
Figure 2. Total renin (A), renin (B) and prorenin (C) levels (n = 16) measured by immunoradiometric assay and angiotensinogen (AGT; D,E) and megalin (F,G) protein abundance (n = 12) measured by Western blot in healthy and early-onset pre-eclamptic (EoPE) placental tissue. Panels (D,F) are representative blots of AGT and megalin protein. Data were normalized versus GAPDH (E) or β-actin (G). Data are mean ± SEM. * p < 0.05, ** p < 0.005, *** p < 0.0005 vs. Healthy. AGT, angiotensinogen.
3. Placental Release of (Pro)renin and AGT
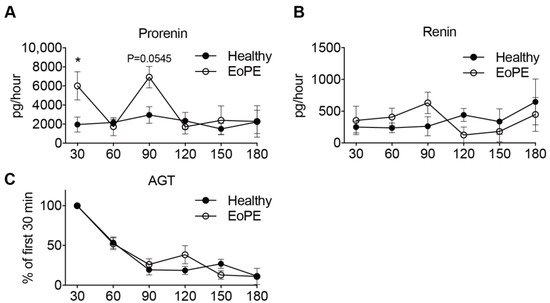
Prorenin release into the maternal perfusate from both healthy (n = 5) and pre-eclamptic (n = 6) placentas remained stable over the entire perfusion period. In two pre-eclamptic placenta perfusions, the perfusion had to be stopped after 90 min because of leakage. Release of prorenin tended to be higher from pre-eclamptic placentas, although significance was reached at two time points only (Figure 3A). Renin release paralleled prorenin release, albeit at >10-fold lower levels (Figure 3B). In contrast, placental AGT release from both healthy and pre-eclamptic placentas peaked initially, and then displayed a steady decline, so that the release was lowest after 180 min (Figure 3C). Neither for renin, nor for AGT, were there differences observed between healthy and pre-eclamptic placentas.
Figure 3. Prorenin (A), renin (B) and angiotensinogen (C) release from isolated perfused healthy (n = 5) and early-onset pre-eclamptic (EoPE, n = 6) cotyledons. AGT: angiotensinogen. Perfusate collection from two of the pre-eclamptic placentas could not be continued over the full 180 min due to fetal-to-maternal leakage. In such cases, only the samples prior to the occurrence of leakage were used. Data are mean ± SEM. * p < 0.05.
4. Megalin Internalizes AGT and PPI Decreases Renin Internalization but Not Binding in BN16 Cells
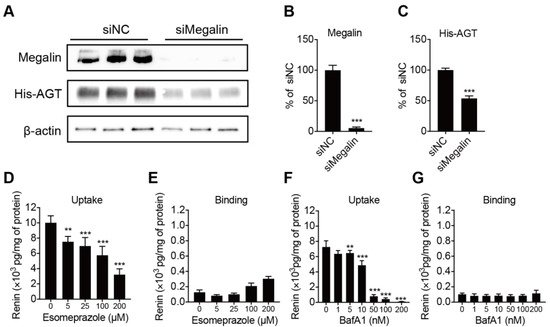
Megalin is known to be a receptor for both renin and prorenin [
27]. To verify whether it also binds and internalizes AGT, we incubated BN16 cells with tagged AGT. BN16 cells accumulated AGT following a 2 h incubation period (
Figure 4A). Inhibiting megalin expression greatly suppressed this accumulation, confirming that megalin also underlies AGT uptake (
Figure 4B,C). We next tested whether the PPI esomeprazole regulates renin binding and internalization. Esomeprazole dose-dependently reduced renin uptake in BN16 cells, but did not affect renin binding (
Figure 4D,E). This fully paralleled the effect of bafilomycin A1 (
Figure 4F,G), and illustrates that esomeprazole, such as bafilomycin A1, interferes with the megalin-mediated internalization process.
Figure 4. Panels (A–C): representative blots of His-AGT protein abundance in BN16 cells treated with negative control (siNC) or siRNA against megalin (siMegalin). Panels, (D–G): cell-associated renin levels after incubating BN16 cells with 100 U/L renin at 37 °C (D,F) or 4 °C (E,G) for 2 h in the presence of increasing concentrations of esomeprazole (D,E) or bafilomycin A1 (F,G). Data are mean ± SEM of n = 5–9. ** p < 0.005, *** p < 0.0005 vs. control.
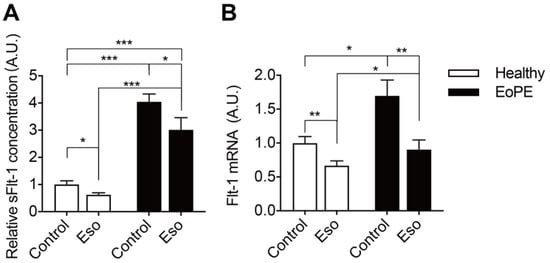
5. PPI Reduces sFlt-1 Secretion in Human Placental Villous Explants
As expected, pre-eclamptic placenta explants release more sFlt-1 than healthy placenta explants (Figure 5A). This is in agreement with the increased Flt-1 expression in pre-eclamptic explants (Figure 5B). Esomeprazole-suppressed Flt-1 expression and sFlt-1 release particularly in pre-eclamptic explants and, as a consequence, Flt-1 expression in pre-eclamptic explants after this drug was identical to that in healthy placenta explants.
Figure 5. sFlt-1 levels in medium (A) and mRNA levels in explants (B) after incubating healthy or pre-eclamptic (EoPE) placental villous explants with 100 μM of esomeprazole (Eso) for 48 h. sFlt-1 level: healthy n = 18, EoPE n = 8; Flt-1 mRNA level: healthy n = 7, EoPE n = 4. * p < 0.05, ** p < 0.005, *** p < 0.0005 vs. 0. A.U., arbitrary unit.
6. Discussion
The existence of a placental RAS has been proposed decades ago [
28]. Although it is believed that it may contribute to pre-eclampsia, its exact role is still unknown. This relates to the fact that we still do not fully understand how local Ang II synthesis occurs in the placenta. In the current study, we found that the mRNA levels of RAS components are comparable in healthy and pre-eclamptic placentas. This agrees with previous studies showing that renin, AGT, ACE and AT2R expression are similar in normal and pre-eclamptic decidua basalis [
15,
29,
30]. Simultaneously, uteroplacental AT1R [
30,
31,
32] and renin [
32] upregulation have also been claimed to be involved in the pathogenesis of pre-eclampsia. One explanation for these conflicting observations is that different tissues were studied, ranging from endometrium and myometrium to decidual tissue. In our study, we collected tissue from the maternal side of the placenta, i.e., decidua basalis and part of chorionic villi. A second explanation is that placental RAS expression may change with increasing gestational age [
32].
Importantly, placental AGT expression in the present study was low and in most cases undetectable. Such low expression was observed earlier, when making a comparison with prolactin and renin using Northern blot analysis [
34]. In situ mRNA detection via the padlock probes method also did not detect AGT expression in the placenta [
35]. Despite the absence of significant expression, AGT protein was easily detectable in placental tissue. This implies that placental AGT must have been taken up from maternal blood. In agreement with this concept, placental AGT levels were lower in pre-eclamptic women, who are known to have greatly diminished circulating AGT levels. Moreover, perfusing the placenta with buffer resulted in a washout of AGT. Animal data similarly support a major role for AGT of maternal origin. When female mice carrying the human renin transgene were crossed with male mice carrying the human AGT transgene (both of which do not react with their mouse counterparts), the pregnant mice remained normotensive. Yet, when crossing female human AGT transgenic mice with male human renin transgenic mice, the pregnant mice did develop a pre-eclamptic phenotype, characterized by hypertension, elevated sFlt-1 levels and proteinuria [
36]. Moreover, specifically inhibiting human AGT expression in the maternal liver with liver-targeted siRNA in the latter model suppressed the pre-eclamptic phenotype [
37]. Taken together, these data imply that placental angiotensin generation relies on maternal AGT.
Unlike AGT release, which peaked immediately and then decreased, placental (pro)renin release remained stable over a 3 h perfusion period in healthy placentas. Since the total renin release in healthy placentas amounted to ≈2000 pg/h, and considering that a cotyledon wet weight amounted to ≈25 g, the total renin amount released during the 3 h perfusion period can be estimated to be 2500/25 × 3 = 300 pg/g. This is 2–3 times the amount detected in placental tissue. These data, therefore, strongly support the continuous synthesis and release of (pro)renin from the placenta, in full agreement with previous findings [
28,
38,
39]. Importantly, the circulating renin levels in pre-eclamptic women are lower than those in healthy pregnant women, while their circulating prorenin levels mimic those in healthy pregnancy [
12]. The present study observed 2-fold higher prorenin levels in pre-eclamptic placentas, while placental renin was identical in healthy and pre-eclamptic women. Placental (pro)renin release paralleled these latter observations. Taken together, these data suggest that placental (pro)renin synthesis occurs independently of renal (pro)renin synthesis. Yet, whether this results in independent upregulation of Ang II generation at placental tissue sites cannot be concluded, particularly since this would rely on the uptake of circulating AGT, which is lower in pre-eclampsia. Here, there might be a role for the megalin/V-ATPase pathway.
The (P)RR is an accessory protein of the V-ATPase. Its upregulation in pre-eclampsia, combined with increased megalin levels, favors increased activity of the megalin–V-ATPase pathway in this condition [
17,
40]. We recently reported that megalin is a novel endocytic receptor for (pro)renin in the kidney [
27]. Inhibition of megalin and the (P)RR similarly suppressed endocytosis, without showing additive effects [
25]. This confirms that megalin-mediated uptake relies on (P)RR/V-ATPase-dependent endosomal acidification. These data were obtained in BN16 cells, which predominantly express megalin, and no other renin/prorenin-binding receptors such as the mannose-6-phosphate receptor [
27]. The latter receptor also occurs in trophoblast cells [
41]. Using megalin-expressing BN16 cells, we were now able to show that the megalin–V-ATPase pathway also underlies AGT uptake. Moreover, we observed that the PPI esomeprazole, such as the V-ATPase inhibitor bafilomycin A1, inhibits megalin-mediated internalization. Given that megalin-mediated AGT uptake underlies renal angiotensin generation [
17], we speculate that a similar phenomenon may occur in the placenta. Here, we emphasize that the greater sensitivity of pre-eclamptic women to Ang II [
11,
42] may imply that placental Ang II levels do not necessarily have to be higher than those in healthy placentas in order to exert the same or even a stronger effect. Onda et al. showed that PPIs lower sFlt-1 synthesis in placental explants (confirmed in the present study), but were unable to link this to a direct consequence of V-ATPase inhibition with bafilomycin A1 [
25]. Our data now reveal a new option: PPIs might suppress placental RAS activity by interfering either with AGT uptake, with (pro)renin transport, or both. Since lower placental Ang II levels are expected to result in diminished sFlt-1 production, this concept offers a novel explanation for the observation that PPIs lower circulating sFlt-1 in pre-eclamptic women (
Figure 6).
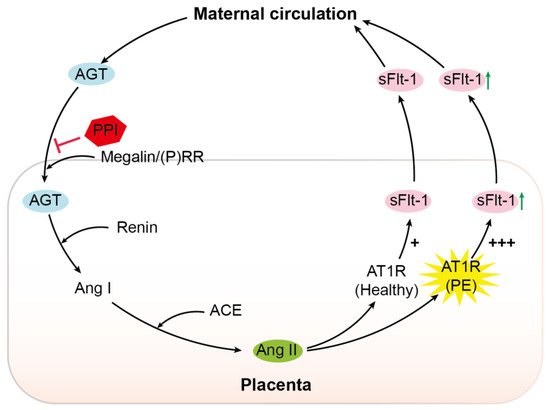
Figure 6. Schematic overview. AGT, angiotensinogen;